1. Introduction
Nanoparticles (NPs) have undergone exponential growth in applications in the biomedical field as drug-delivery vehicles, high-contrast imaging agents, or active therapeutics [
1]. In particular, in in vivo imaging NPs offer many advantages when compared to molecular imaging agents, such as greatly improved signal-to-noise ratios, stable signal generation, high spectral resolution for multiplexed detection, and the possibility of multimodal signal generation.
As a result, NPs have been proposed for the imaging of cancer, in a range of modalities which include near-infrared (NIR) fluorescence, magnetic resonance imaging (MRI), and positron emission tomography (PET). Indeed, NPs accumulate preferentially in tumor tissues in comparison with the normal ones. This is mainly because the tumors have impaired lymphatic drainage, enhanced permeation, and retention (EPR).
Very often, unfunctionalized, bare NPs tend to fail when challenged in the human body [
2]. The cell uptake of naked particles can be very limited, because the cell membrane is a selectively permeable barrier, which efficiently protects the cell interior from the environment. In addition, as soon as NPs are introduced into a complex biological environment, a mixture of proteins interacts with the particle surface (protein corona), influencing the pharmacokinetic properties and limiting the effect of the particle. For instance, the adsorption of the opsonins plasma proteins (such as immunoglobulin G, fibrinogen, complement factors) to the NP surface plays a prominent role in triggering clearance via phagocytosis.
On the other hand, NP–cell interactions can be greatly improved by modifying their surface. A proper surface coating can tune the magnetic, electrical, chemical, and optical properties, which affect pharmacokinetics, distribution, accumulation, and toxicity. Stealth polymers such as polyethylene glycol (PEG) are widely utilized to limit opsonization and NP clearance from the circulation. These coatings form a protective layer around the NP, neutralizing surface charge, conferring hydrophilicity, and providing a steric barrier to adsorption. PEG coating is also known to promote more efficient hepatobiliary clearance.
In this respect, peptide coating has emerged as a potent strategy for influencing NP behavior in vivo [
3]. Peptides can modify cell uptake and biological stability and decrease susceptibility to clearance of the NPs. For instance, previously we designed peptide-functionalized nanosystems mimicking the pro-apoptotic Smac/DIABLO mitochondrial protein. To the purpose, inorganic fluorescent NPs coated with a biocompatible organic shell were directly functionalized by [3+2] alkyne-azides cycloaddition with an Smac/DIABLO-derived peptide and/or a tumor-homing RGD integrin ligand peptide. At low micromolar concentration, these NPs showed significant toxicity towards several cancer cell lines, correlated with high stability and bioavailability, and increased levels of apoptotic activity, while being much less toxic towards model healthy cells [
4].
As a consequence, several chemoselective reactions have been optimized for anchoring peptide sequences onto the surface of the NPs, e.g., the [3+2] cycloaddition between azides and strained or terminal alkynes, olefin metathesis, maleimide-thiol reaction, etc. [
5].
In this work we propose an alternative procedure for the preparation of peptide-functionalized NPs made up of self-assembling surfactant tri-block copolymers with a fluorescent silica core. We opted for silica because it is a biocompatible, versatile material for the fabrication of NPs for theragnostic applications [
6]. It is hydrophilic and stable in physiological conditions and inert from a photophysical point of view [
7]. Fluorescent dyes can be easily inserted into a silica matrix by covalent linking and this embedding usually provides a very bright luminescence signal due to the accumulation and to the increased luminescence properties of the dyes. Silica chemistry is also well established, offering many possibilities for surface functionalization schemes.
The micellar NPs have been obtained from peptide-block copolymers [
8], prepared by conjugation of the peptides to block copolymers by copper-catalyzed azide-alkyne chemistry (CuAAC, click chemistry), prior to NP formation. In fact, in our experience the coupling yield between peptides and preformed copolymer NPs can be disappointing. This could be due to several reasons, such as (i) a scarce accessibility of functional groups on the NP surface, which depending on their polarity can be buried within the outer polymeric layer, (ii) the slow diffusion of NPs compared to molecular reagents, and finally (iii) the necessity to perform the coupling in aqueous media. Even more, with a post-functionalization process it is impossible to verify coupling efficacy by direct analytical methods, such as NMR spectroscopy, but just with indirect fluorometric or colorimetric methods that can be affected by the presence of some interferences.
2. Materials and Methods
2.1. General Methods
Chemical reagents, including protected amino acids, were purchased from commercial sources and used without purification.
Peptide purity was assessed by analytical RP HPLC performed on an 1100 series apparatus Agilent Technologies, Milan, Italy, using an XSelect Peptide CSH C18 column (Waters, Milford, MA, USA), 4.6 mm × 100 mm, 130 Å, 3.5 μm. The mobile phase was a mixture of 0.5% HCOOH in H2O and 0.5% HCOOH in CH3CN for ionic peptides (method A) or H2O/CH3CN for neutral peptides (method B). Alternative to HPLC-MS, purities were assessed by reverse-phase ultra-performance liquid chromatography (RP-UPLC), using a reverse-phase (RP) column mod. Acquity UPLC ®BEH C18 1.7 μm (2.1 × 50 mm); DAD 210 nm; DAD 254 nm; mobile phase: from 2:8 solvent A/solvent B to 7:3 solvent A/solvent B, in 26 min, at a flow rate of 0.3 mL/min, followed by 4 min at the same composition; solvent A = 0.1% HCOOH in H2O, B = 0.1% HCOOH in CH3CN.
MS (ESI) analysis was performed using an MS single quadrupole HP 1100 MSD detector (Agilent Technologies, Milan, Italy).
Fluorescence measurements were performed with an LS-55 Fluorescence Spectrometer (Perkin Elmer, Milan, Italy).
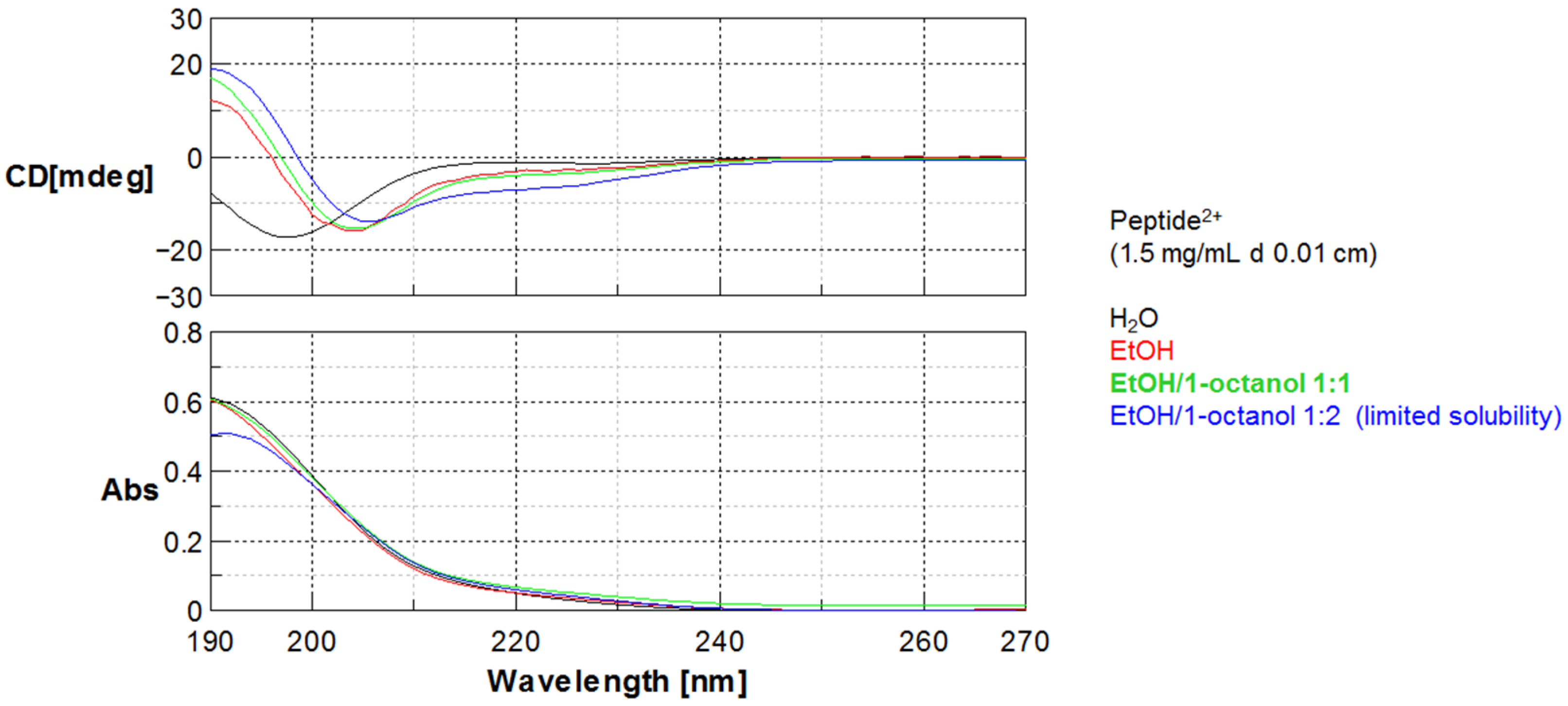
Circular dichroism (CD) measurements were performed at room temperature with a Jasco J-715 spectropolarimeter by using a circular quartz cell of 0.1 mm path length. Reported CD spectra were obtained by taking the average of five scans made at 100 nm/min. Spectral data are expressed in units of millidegree.
Fourier transform IR (FT IR) analysis was performed on an Alpha FT IR spectrophotometer (Bruker, Billerica, MA, USA).
1H NMR was performed at 400 MHz on a Varian Gemini 400 (Agilent) in 5 mm tubes in CDCl3 or DMSO-d6 at RT; chemical shifts are reported as δ values relative to residual CHCl3 (δH = 7.26 ppm) or to residual DMSO (δH = 2.50 ppm). In general, CDCl3 was used for the derivatives of PF127 and DMSO-d6 for the chimeras.
Dynamic Light Scattering (DLS) for the determination of the hydrodynamic diameter distribution of the NPs was carried out employing a Malvern Nano ZS instrument equipped with a 630 nm laser diode. Samples were housed in disposable polystyrene cuvettes of 1 cm optical path length. Samples were prepared diluting 300 μL of NP solution in 900 μL of water and 60 μL of PBS 10× (Phosphate-buffered saline, pH 7.4, 1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4).
Zeta Potential was measured with the same instrument; the samples were prepared by diluting 100 μL of NPs solution, 120 μL of 0.02 M KCl, 120 μL of 0.1 M EPPS (3-[4-(2-Hydroxyethyl)piperazin-1-yl]propane-1-sulfonic acid, pH 8) in 860 μL of water.
Transmission electron microscopy (TEM) was conducted in a Philips CM 100 TEM operating at 80 kV. For the investigations, conventional Formvar-film copper microgrids were dried under vacuum after deposition of a drop of NP solution diluted with water (1:50). The size distribution was obtained by analyzing the images manually using the software ImageJ (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA).
Confocal images were obtained with a C1s confocal laser-scanning microscope equipped with a PlanApo 60× or 40× oil immersion lens (Nikon, Tokyo, Japan).
2.2. Peptide Synthesis
General procedure. Fmoc-Rink Amide resin (0.5 g, substitution 0.45 mmol/g) was swollen in DMF (5 mL) for 15 min before the use. Fmoc deprotection was carried out by treatment with 20% piperidine/DMF (5 mL) for 20 min; after filtration, the treatment was repeated for further 40 min. The resin was subsequently washed 3 times with DMF, Et2O, DCM (5 mL each). Fmoc-amino acid (2.5 eq.) carrying orthogonal protecting groups at the side chains and HOBt (2.5 eq.) were dissolved in DMF (3 mL), and after 10 min this mixture was added to the resin at RT. Then, TBTU (2.5 eq.) and DIPEA (4.0 eq.) were also added and the mixture was shaken for 3 h at RT. The suspension was filtered, and the resin was subsequently washed 3 times with DMF, Et2O, DCM (5 mL each). Coupling efficacy was monitored by the Kaiser test. The final coupling with 5-hexynoic acid was carried out in the presence of HOBt/TBTU/DIPEA under the same conditions as described above. The residues Lys, Ser, Asp, Gln, Glu, were introduced as Fmoc-Lys(Boc)-OH, Fmoc-Ser(tBu)-OH Fmoc-Asp(tBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(tBu)-OH, respectively. The removal of the acid-labile groups at the side chains occurred during the cleavage of the sequences from the resin.
The cleavage of the peptide from the resin was performed by treatment with TFA/TIPS/H2O/PhOH (88:5:5:2 v/v/v/m, 10 mL) while shaking for 6 h at RT. The mixture was filtered, and the resin was washed in sequence with 1% TFA in Et2O (5 mL), DCM (5 mL), and MeOH (5 mL). The filtrate and the washes were collected, and the organic solvents were removed under reduced pressure. The resulting residue was suspended in ice-cold Et2O, and the crude solid that precipitated was collected by centrifugation (70–80% yield, 85–95% pure as determined by RP HPLC).
Hex-5-ynamide-Lys-Ala-Leu-Ser-Lys-Leu-Leu-NH2 (peptide2+) was isolated as a beige solid as a 2xTFA salt (385 mg, 78% yield compared to the resin load). Purity was determined to be 94% by analytical RP HPLC (method A), Rt = 1.98 min. MS (ESI) m/z: calculated 865.58 [M+H]+; found 433.8 [M+2H]2+.
Hex-5-ynamide-Ala-Aib-Ala-Aib-Ala-Gly-NH2 (peptide0) was isolated as a pale yellow waxy solid (92 mg, 82% yield compared to the resin load). Purity was determined to be 78% by analytical RP HPLC (method B), Rt = 5.8 min. MS (ESI) m/z: calculated 551.3 [M+H]+; found 552.8 [M+H]+
Hex-5-ynamide-Phe-Pro-Glu-Leu-Gln-Gln-Asp-NH2 (peptide2−) was obtained as a beige solid (73 mg, 67% yield compared to the resin load). Purity was determined to be 90% according to analytical RP UPLC, Rt = 3.6 min. MS (ESI) m/z: calculated 969.5 [M+H]+; found: 969.3 [M+H]+, 991.3 [M+Na]+, 485.3 [M+2H]2+.
2.3. Synthesis of Polymer-(N3)2 and Polymer–Peptide Conjugation
Pluronic® F127 (6.3 g, 0.5 mmol, 1 eq.) was dried by azeotropic distillation with toluene at reduced pressure. Then, the polymer was solubilized in anhydrous DCM (25 mL) and treated with triethylamine (TEA, 140 μL, 1 mmol, 2 eq.) and methanesulfonyl chloride (MsCl, 77 μL, 1 mmol, 2 eq.) under inert atmosphere. The mixture was stirred at 0 °C for 3 h and then at RT overnight. The solvent was evaporated at reduced pressure, the resulting residue was washed with ice-cold Et2O, and solid was collected by precipitation (>95%).
Sodium azide (122 mg, 1.88 mmol, 4 eq.) was added to a suspension of dimesylate–PF127 (6.026 g, 0.47 mmol, 1.0 eq.) in CH3CN (25 mL), and the mixture was stirred under reflux for 48 h. Subsequently, the solvent was removed under reduced pressure. The solid so obtained was dispersed in a 5% NaHCO3 solution saturated with NaCl (20 mL). The mixture was extracted four times with DCM (4 × 15 mL). The combined organic phases were dried over Na2SO4, filtered, and evaporated at reduced pressure, affording a white solid (95%).
PF127-(N3)2 (450 mg, 1 eq) was dissolved in dry CH3CN, and alkyne-peptide (2 eq.), 2,6-lutidine (4 eq), DIPEA (4 eq), and CuI (0.2 eq), were added under an inert atmosphere. The mixture was stirred at RT overnight. Then, the mixture was evaporated at reduced pressure and the solid was dispersed in a 1 M HCl solution saturated with NaCl. The mixture was extracted three times with AcOEt (10 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated at reduced pressure. The resulting solid was washed with ice-cold Et2O, and the solid was collected by precipitation (90%).
2.4. Preparation of the Dye-Loaded NPs
A mixture of pristine PF127 with 10%, 20%, or 30% in weight of polymer–(peptide)2 was dispersed in 3.2 mL of a solvent mixture composed of DMF (38.7% v/v) in 1 M of AcOH containing 0.85 M NaCl. Then, an 8.67 mg/mL solution of rhodamine B triethoxysilane (RhB-TES) in MeOH (300 μL) was added. The mixture was stirred for 1 h, then TEOS (350 μL) was added. After 3 h under stirring, TMSCl was added (40 μL), and the reaction was stirred for 48 h at RT. The NPs were purified by dialysis in a cellulose dialysis tube (Sigma, MW cutoff >12 kDa, avg. diameter 33 mm) against bi-distilled water at RT. The procedure gave NPs prepared with a content of polymer–peptide of 10, 20, and 30%, in short NP-10%peptide, NP-20%peptide, NP-30%peptide, respectively.
2.5. Cells and Culture Conditions
Cells were purchased from the European Collection of Authenticated Cell Cultures ECACC. The cell lines were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin-streptomycin (10,000 U/mL). The cells were grown at 37 °C in a controlled atmosphere (5% CO2/95% air). Confluent cells were split by trypsinization and used at the third or fourth passage after thawing. For all the experiments, the cells were plated at a density of 10,000 cells/cm2. All the reagents for cell culture were purchased from Sigma-Merck (Darmstadt, Germany).
2.6. Cell Viability Assays
Caco-2 cells were plated in 96 multi-well plates in the growth medium (10,000 cells/100 μL per well). Then, the cells were treated with cell culture medium containing the NPs for 24 and 48 h. At the end of treatments, 20 μL of MTS reagent (CellTiter 96 AQueous One Solution Cell Proliferation Assay, (Promega, Madison, WI, USA) was added to each well. After 3 h incubation in the cell culture incubator, the absorbance was read in a multi-well plate reader at 450 nm. Eight wells were used for each experimental point and each independent experiment was repeated three times. The cell viability results were normalized to time-point-matched controls; NP stock solution (200 mM in PBS) was diluted in the growth medium and added to the wells. In control wells only the growth medium was added.
2.7. In Vitro Cellular Uptake Study
The cellular uptake of NP-30%peptide2+ into A549 cells was studied using confocal microscopy. Cells were grown on sterile glass coverslips for 48 h and then treated with 1 μM peptide–NPs for 1 h. The cells were washed with PBS (3×) and fixed in 500 μL of 3% paraformaldehyde. The glass slides were washed twice with 1 mL of 0.1 M Gly in PBS and washed twice again with 1 mL of 1% BSA in PBS. The samples were first incubated with mouse anti-α-tubulin primary antibody for 1 h in agitation at RT. The samples were washed again twice with 1 mL of 1% BSA in PBS and then the cells were stained with Hoechst33342. The specimens were embedded in Mowiol and analyzed by confocal microscopy. The visualization and quantification of cells that internalized the rhodamine B (RhB)–NPs were performed using ImageJ (NIH, Bethesda, MD, USA).
4. Discussion
During the last decades, several NPs have been designed and evaluated for their potential biomedical applications [
15]. Although NPs are frequently proposed as diagnostic agents, only iron oxide NPs have found their way into the clinical routine so far [
16]. This fact is primarily originated by difficulties in achieving appropriate pharmacokinetic properties and reproducible synthesis of monodispersed NPs. In addition, suspicions exist regarding their biodegradation, elimination, and toxicity.
In particular, the cell uptake of the particles can be very limited. In principle, NPs can take advantage of different internalization mechanisms, i.e., phagocytosis, pinocytosis, clathrin- or caveolae-dependent endocytosis, and direct penetration, and the preference for one mechanism over the others is highly dependent on the precise NP characteristics. The situation is further complicated by the possibility of particles to exploit different pathways at the same time. Even when NPs have penetrated cells, the large majority of the particles may be entrapped in endosomes and organelles.
The most frequently utilized NP formats fall under one of the following categories: gold nanoparticles (AuNPs), magnetic nanoparticles (MNPs), semi-conducting NPs and quantum dots (QDs), mesoporous silica nanoparticles (MSNPs), liposomes, and polymer nanoparticles. The latter are highly appreciable, since the versatility of synthetic polymers makes possible a wide range of particle architectures. Amphiphilic block copolymers are particularly appealing since they tend to self-assemble into micelles whose size, membrane thickness, porosity, and other features can be tuned by adjusting block length and pendant functional groups.
Previously, we described micelles formed by the self-assembly of the tri-block surfactant copolymer Pluronic
® F127 (PF127) (polyethylene glycol–polypropyleneoxide–polyethylene glycol, PEG
100–PPO
65–PEG
100) [
17]. This block copolymer alone can form micelles in aqueous media with a hydrophobic core, which can be used to non-covalently encapsulate dyes, cargoes, or drugs [
18,
19].
However, polymeric self-assembled structures are dynamic systems, and their stability is influenced by their local concentration, pH, and by the presence of other molecules or proteins [
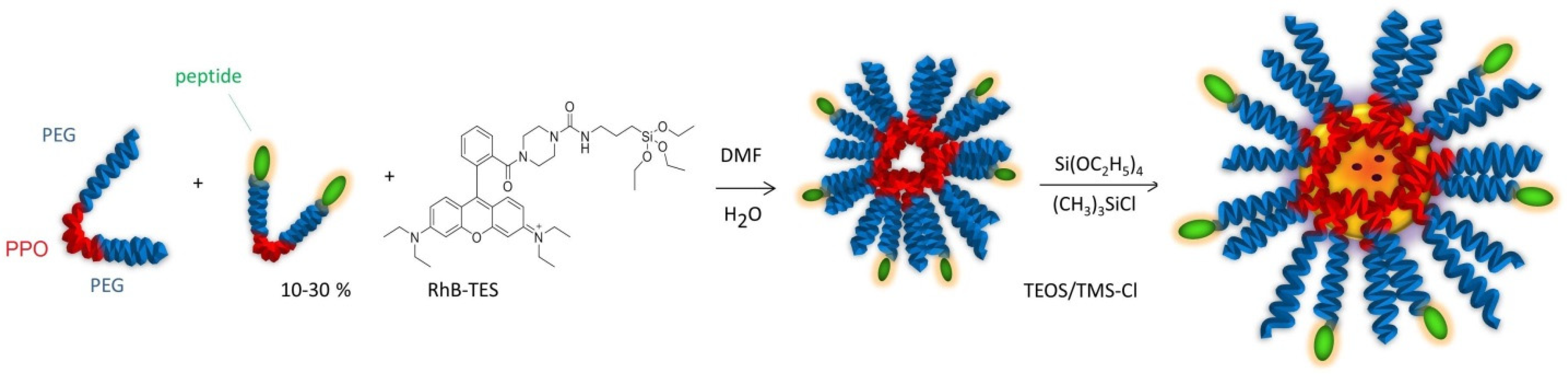
20]. To increase the stability, the hydrophobic core of the micelles was trapped into a silica frame by polymerization of TEOS/TMSCl, giving silica-core/PEG-shell NPs (
Figure 6).
These particles combine the versatility of the micelles with the stability of the silica NPs. Silica is an intrinsically non-toxic material and is photophysically inert, but it can be easily doped with a variety of dyes to achieve the desired luminescence emission properties [
21]. The dye rhodamine B, triethoxysilane (RhB-TES) was covalently linked to the silica matrix to avoid leaching of the dye; this inclusion also increased the photostability of the dye [
7].
The micelles formed by the Pluronic copolymer present on the surface a shell of PEG. This hydrophilic polymer is expected to increase NP colloidal stability in physiological conditions, reduces non-specific binding, and opposes the uptake by the reticuloendothelial system [
22]. However, there is a fine line between providing a coating that improves biodistribution and diminishing interactions with target cells and tissues. The coating of NPs with PEG can reduce cellular uptake and therapeutic efficacy [
23], the generation of PEG-specific antibodies which accelerate clearance [
24], and preferential accumulation in the liver and spleen [
25].
In this context, we became interested in the preparation of fluorescent silica-core/PEG-shell NPs functionalized with diverse peptide sequences. Indeed, peptide coating is currently regarded as a potent strategy for improving cell uptake and stability and decreasing clearance of the NPs. Compared to full-size proteins, peptides offer several advantages, being generally less expensive, more stable, and less immunogenic. Due to their small size, peptides do not alter to a significant extent the NP size.
As a consequence, NPs have been decorated with a variety of peptides [
3,
4,
15]. Among them, the cell-penetrating peptides (CPPs) are positively charged sequences that generally consent higher cell penetration, thanks to electrostatic interactions with negatively charged phospholipids. This is followed by destabilization of the cell membrane, for example through a pore or inverted micelle formation. Yet, CPPs may indiscriminately cross cell membranes, leading to the non-selective uptake of NPs by almost all kinds of cells [
26].
Alternatively, NPs can be functionalized with peptide sequences that specifically bind membrane receptors or other exposed biomolecules to trigger receptor-mediated endocytosis. For instance, the tumor-targeting precision of NPs [
5] or nanostructured materials [
27,
28,
29] can be greatly improved by conjugation with peptide ligands targeting the integrin receptors overexpressed by cancer cells.
Apart from cell entry, peptides can influence other pharmacokinetic aspects. The formation of the protein corona can be modulated by promoting the absorption of dysopsonins. The peptide CD47 was used to inhibit phagocytosis. In addition, many peptide sequences have been identified that can promote endosome escape after endocytosis of NPs [
1,
2,
3,
4].
Several reactions are currently utilized for site-selective grafting of the peptide without perturbing the remaining amino acids [
30,
31], e.g., amine-
N-hydroxysuccinamide coupling, Michael addition, CuAAC, Staudinger ligation, oxime bond arrangement, thiazolidine ring formation, palladium-catalyzed couplings [
32], urea from isocyanates [
33], etc. These reactions proceed with variable degrees of efficiency [
4,
26], giving more or less homogenous peptide coatings.
In this work, we opted for an alternative approach to the preparation of peptide-functionalized silica-core/PEG-shell NPs. Indeed, the diazide derivative of the tri-block copolymer PF127, obtained in turn from the dimesylated derivative, was functionalized by CuAAC with alkyne-peptides at an early stage of the synthetic pathway (
Figure 3).
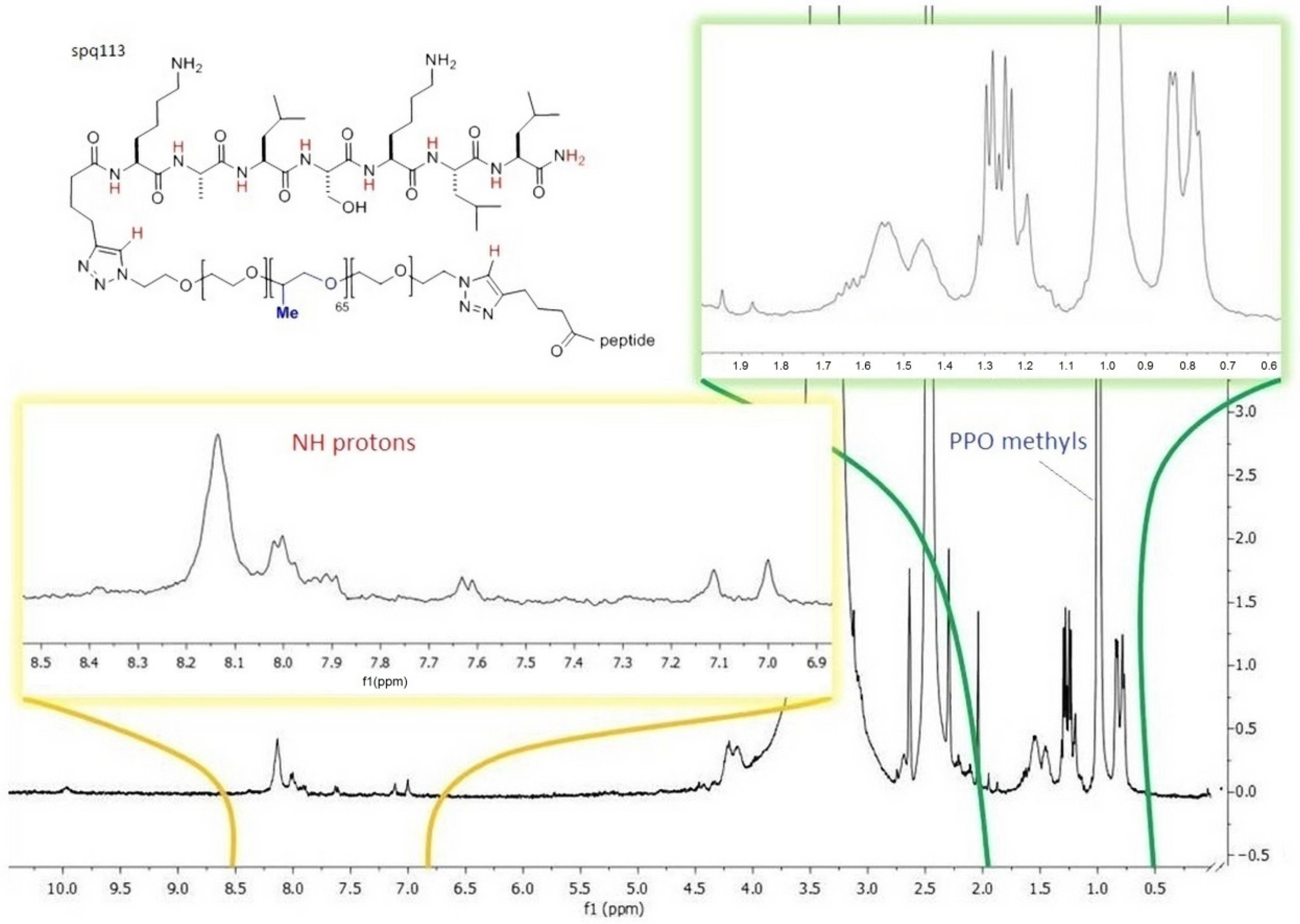
The subsequent derivatizations of PF127 and the conjugation with the peptides were monitored by
1H NMR and FT IR (
Figure 4 and
Figure 5), which confirmed that each step proceeded in almost quantitative yield, and confirmed the reproducibility of the procedures [
34,
35].
To control any effects of peptide charge in the formation of polymeric micelles [
34,
35], we utilized a model peptide with net 2+ positive charge (peptide
2+), a neutral peptide (peptide
0), and finally a peptide with a net 2- negative charge (peptide
2−). The 2+ sequence was derived from bioactive peptide PTP7 [
36], the neutral sequence was derived from the highly folded, 3
10-helical foldamer Cbz-(Ala-Aib)
2-Ala-Gly-NH
2 [
37], while the third 2- anionic sequence was derived from a peptide isolated from frog skin, Pleurain-C2 (
Figure 1) [
38].
In addition to the net charge, peptides slightly differ for the folding behavior, as evidenced by CD data described above. In particular, for peptide0 the same helicity is expressed in ethanol or 1-octanol while for peptide2+ in analogous solvent conditions the helical folding is influenced by solvent polarity.
Thereafter, the resulting palindromic peptide–polymer block copolymers were utilized for NP formation (
Figure 6). Blend and mixed peptide–NPs were also prepared to the aim of understanding whether possible combinations between positively and negatively charged peptides might influence the formation and the properties of the micelles. These NPs present on their surface diverse combinations of positive and negative peptides. The use of NPs with non-ionic peptides, or NPs with zwitterionic balanced charge, may be particularly useful for reducing clearance [
3].
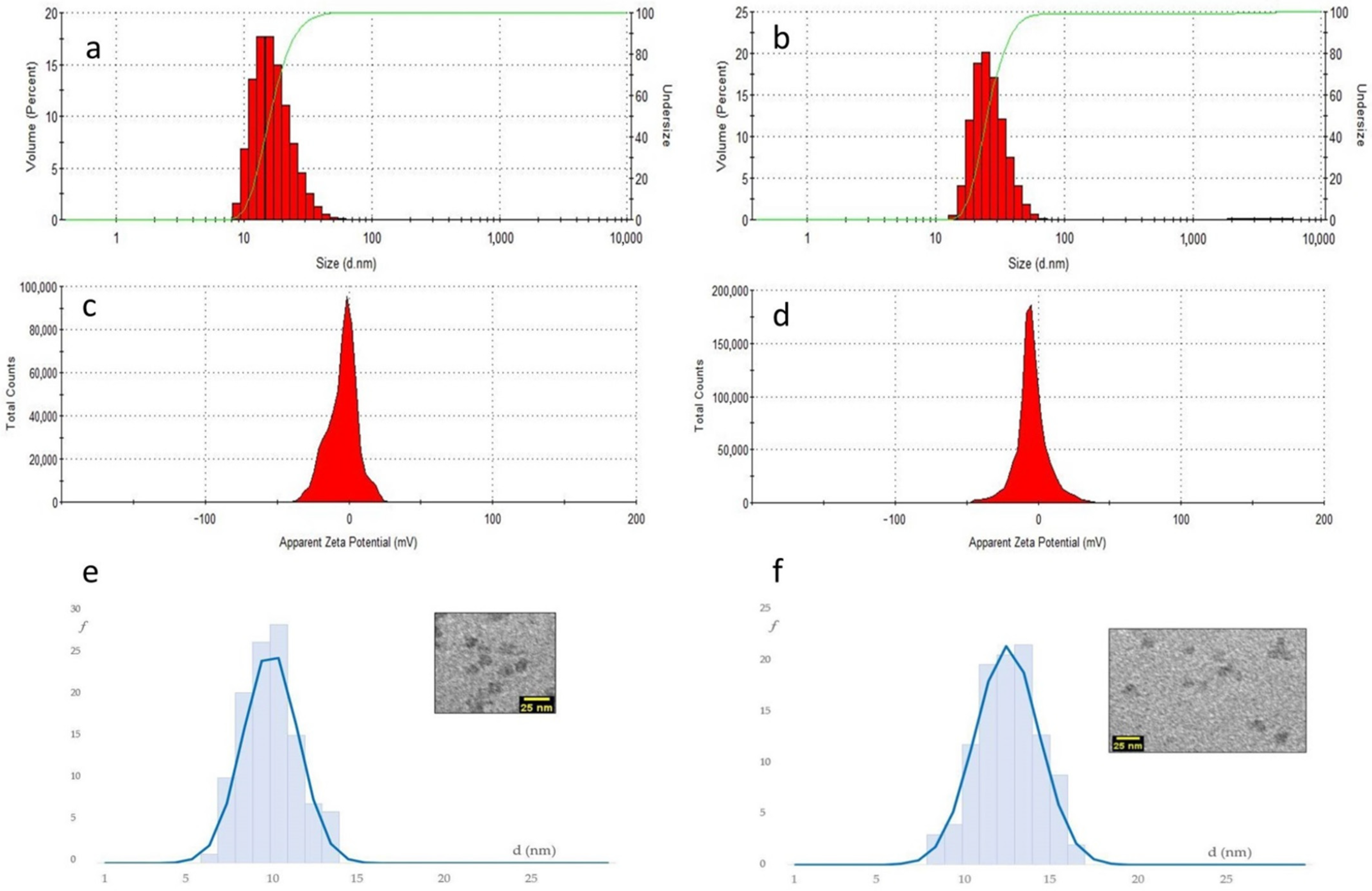
As it turned out, DLS revealed that the diverse peptide–NPs showed much higher hydrodynamic diameters as compared to bare silica-core/PEG-shell NPs (around 22 nm), and the values increased with the increase in peptide functionalization, up to 35 ± 1 nm for NP-30%peptide
2+ (
Figure 7, and
Table 1).
By far, the highest hydrodynamic diameters were observed for blend and mixed NPs, which also contained 30% of Pluronic-peptide, >41 ± 1 nm. This can be reasonably associated in part to the mere presence of the peptide, and in part to the solvation of the charged side chains. Indeed, for NPs carrying the neutral peptide0, the increase in dh was inferior, up to 31.5 ± 0.5 nm for NP-30%peptide0.
The NP prepared with the anionic peptide revealed very large dh values, plausibly due to the formation of aggregates (
Table 1). NP aggregation and agglomeration have been recognized to affect cellular uptake and even induce potential toxicity based on the nanoparticle composition and the cell type.
The diameter of the silica core (d
core) of the peptide–NPs was measured by TEM (
Figure 7,
Table 1, and
Supplementary Materials). The composition and the charge of the peptide grafted onto the NPs had a scarce impact on the size of the silica cores. The d
core values were still very close to the bare NPs, 10 ± 1 nm. Only the NP with 30%peptide showed a moderate increase to 13 ± 2 nm. Hence, the larger hydrodynamic diameters measured by DLS plausibly depend on the solvation of the peptide coating and on dynamic interactions between NPs.
The Zeta Potential values (ζ) determined at pH 8 were in the −3 ÷ −5 mV range, and very similar for all samples. It is well known that bare silica NPs are characterized by very negative Zeta Potential values (<20 mV). In contrast, silica-core/PEG-shell NPs have almost neutral Zeta Potential values, indicating that the silica core is buried within a compact polymer shell that efficiently shields from the solvation of charged chemical species.
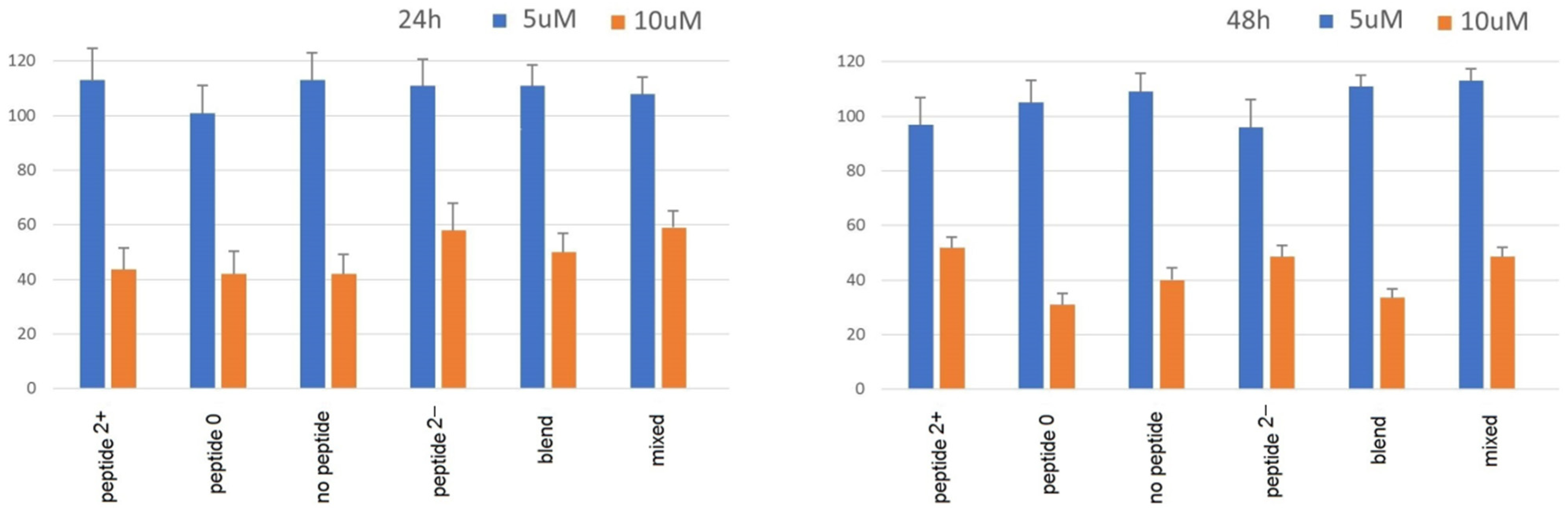
Despite the PEG shell and peptide coating, toxicity of these NPs can still represent a fundamental issue for potential diagnostic applications in vivo. Peptides themselves can induce cytotoxicity, in particular amphipathic peptides which induce membrane damages [
26]. Therefore, the cytotoxicity of all nanoparticles was estimated in vitro in model Caco-2 cells by the MTS cell viability assay (
Figure 8).
For all NPs, the experiments showed no toxicity after 24 or 48 h at the concentration of 5 μm, while a significant loss of vitality was observed at the concentration of 10 μm. Nevertheless, for the NPs carrying the anionic peptides the cytotoxicity in vivo cannot be excluded. Indeed, agglomerated NPs have drastically altered properties. Aggregated particles display a greatly decreased surface area and reduced cellular uptake, which can lead to an underestimation of toxicity before application in vivo.
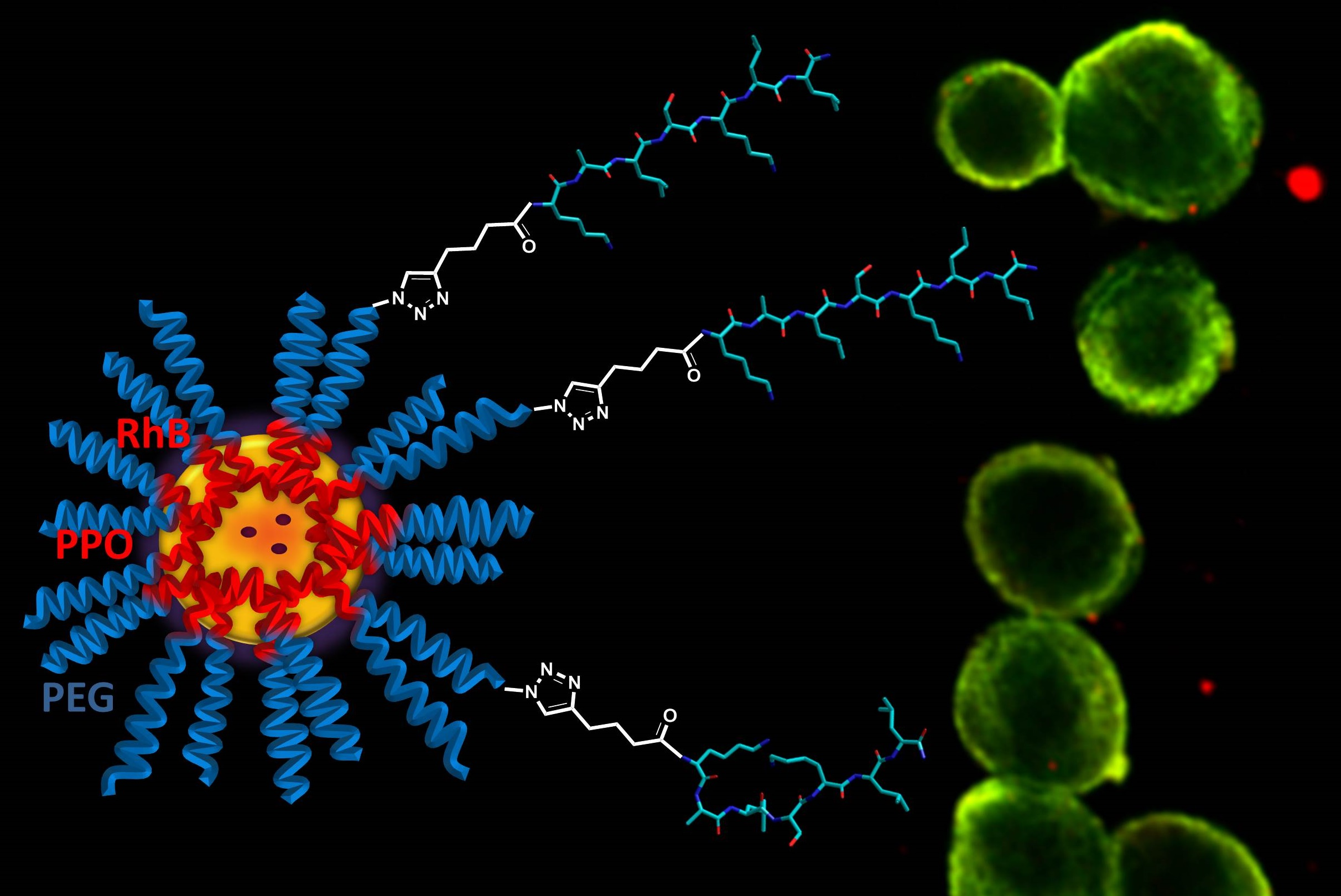
Finally, the cell penetration ability of the fluorescent peptide-coated silica/PEG NPs was preliminarily investigated in model A549 cells. As a proof of concept, the internalization of the NP-30%peptide
2+ was followed by confocal microscopy (
Figure 9). The images confirm a significant internalization. The merging of the images of the cytoskeleton (green) with the NPs (red) showed orange localizations which suggest that the NPs are bound to tubulin.
At present, the definition of the precise mechanism by which these NPs enter the A549 cells is beyond the scope of this work. In general, it is known that cationic sequences can first strongly bind negatively charged cell membranes and then induce transport via either a direct or endocytic pathway. However, other mechanisms, e.g., receptor-mediated, cannot be ruled out.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}