
Solute-Induced Perturbation of the Solvent Microstructure in Aqueous Electrolyte Solutions: Some Uses and Misuses of Structure Making/Breaking Criteria


Abstract
:1. Introduction
2. Fundamentals Underlying the Description of the Microstructural Perturbation of the Solvent Environment upon Solute Solvation
2.1. What Does Really Mean That a Solute Strengthen/Weaken the Structure of the Solvent?
2.2. Need to Provide an Explicit Definition/Criterion for the Structure Making/Breaking Ability of a Solute Species
3. Critical Analysis of the Ambiguity of Two Widespread Structure Making/Breaking Markers
3.1. Why Hepler’s Isobaric-Thermal Expansivity Criterion Cannot Describe a Structure Making/Breaking Event?
3.2. Why the Behavior of the Jones–Dole’s B-Coefficient Cannot Be Taken as a Structure Making/Breaking Marker?
4. Experimental Evidence of Structure Making/Breaking Behavior in Aqueous Electrolytes and Comparison against Predictions from the Conjectured Markers
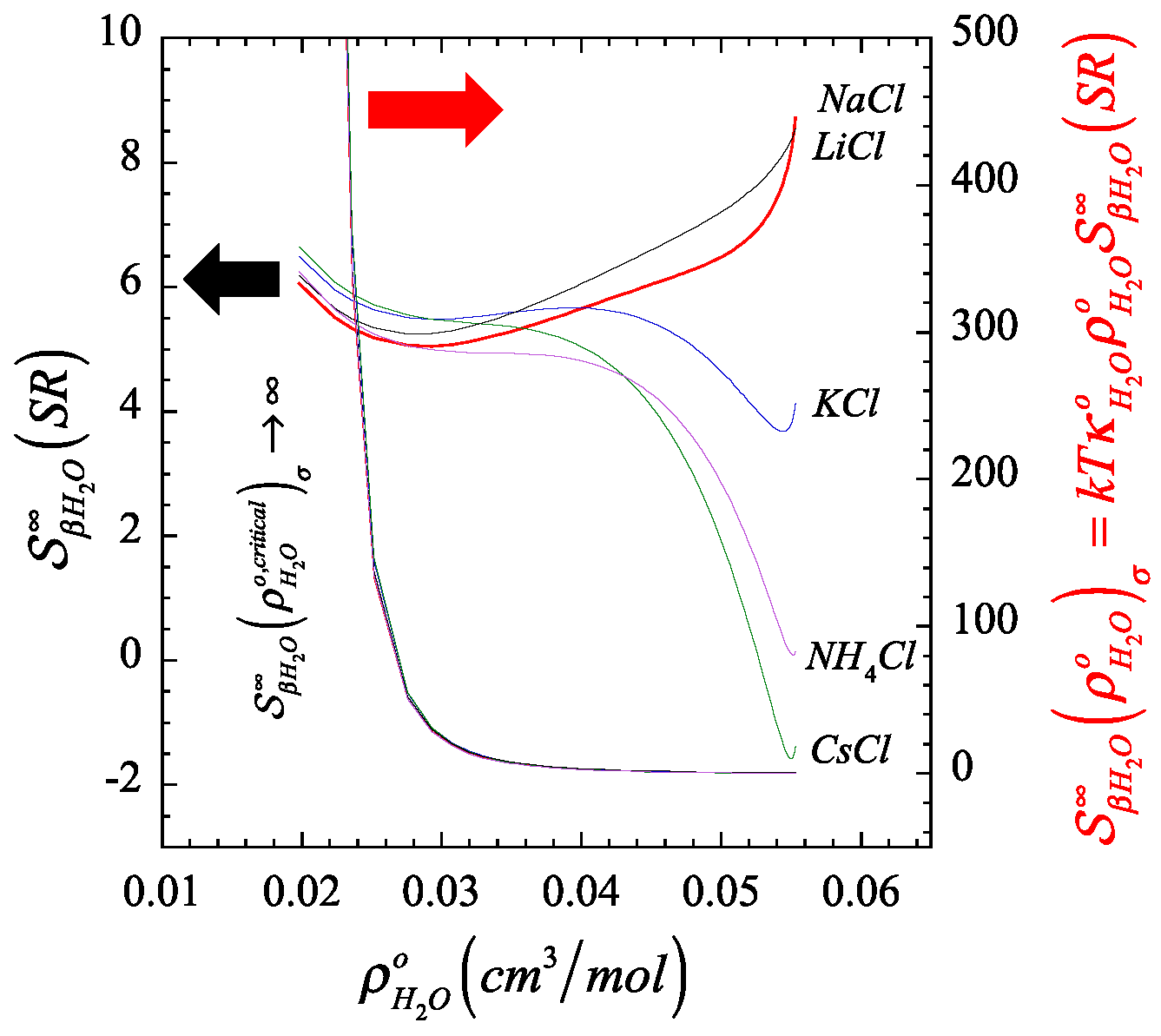
4.1. Illustration of the Behavior of the Short- and Long-Range Contributions to the Structure Parameter along the Liquid Branch of the Coexistence Phase Envelope of Water
4.2. Comparison between the Predictions of the Two Common Structure Making/Breaking Markers and the Actual Microstructural Behavior
5. Final Remarks and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Transition State Interpretation of Jones–Dole’s B-Coefficient
Appendix B. Structure Making/Breaking Parameter from the SOCW Representation of the Partial Molar Volumes of Simple Electrolyte Solutes
Appendix C. Practical Guide to the Calculation of the Fundamentally-Based Structure Making/Breaking Marker
- Calculate the partial molar volume of the β–solute at infinite dilution as the composition limiting behavior ;
- Calculate the partial molar volume of the pure solvent, i.e., ;
- Calculate the structure making/breaking parameter , Equation (7), after considering the stoichiometric parameter of the solute, either for an ionic solute or for a non-dissociative solute;
- Compare the ratio with the stoichiometric parameter:
- If , then , i.e., the solute behaves as a structure making at the prevailing state conditions;
- If , then , i.e., the solute behaves as a structure breaking at the prevailing state conditions;
- If , then , i.e., the solute induces a negligible structure perturbation at the prevailing state conditions.
References
- Debenedetti, P.G.; Mohamed, R.S. Attractive, Weakly Attractive and Repulsive Near-Critical Systems. J. Chem. Phys. 1989, 90, 4528–4536. [Google Scholar] [CrossRef]
- Chialvo, A.A. Solvation Phenomena in Dilute Solutions: Formal, Experimental Evidence, and Modeling Implications. In Fluctuation Theory of Solutions: Applications in Chemistry, Chemical Engineering and Biophysics, 1st ed.; Matteoli, E., O’Connell, J.P., Smith, P.E., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 191–224. [Google Scholar]
- Chialvo, A.A. On the Solvation Thermodynamics Involving Species with Large Intermolecular Asymmetries: A Rigorous Molecular-Based Approach to Simple Systems with Unconventionally Complex Behaviors. J. Phys. Chem. B 2020, 124, 7879–7896. [Google Scholar] [CrossRef] [PubMed]
- Fulton, J.L.; Heald, S.M.; Badyal, Y.S.; Simonson, J.M. Understanding the Effects of Concentration on the Solvation Structure of Ca2+ in Aqueous Solution. I. The Perspective on Local Structure from EXAFS and XANES. J. Phys. Chem. A 2003, 107, 4688–4696. [Google Scholar] [CrossRef]
- D’Angelo, P.; Roscioni, O.M.; Chillemi, G.; Della Longa, S.; Benfatto, M. Detection of second hydration shells in ionic solutions by XANES: Computed spectra for Ni2+ in water based on molecular dynamics. J. Am. Chem. Soc. 2006, 128, 1853–1858. [Google Scholar] [CrossRef]
- Antalek, M.; Pace, E.; Hedman, B.; Hodgson, K.O.; Chillemi, G.; Benfatto, M.; Sarangi, R.; Frank, P. Solvation structure of the halides from x-ray absorption spectroscopy. J. Chem. Phys. 2016, 145, 044318. [Google Scholar] [CrossRef] [Green Version]
- Zitolo, A.; Chillemi, G.; D’Angelo, P. X-ray Absorption Study of the Solvation Structure of Cu2+ in Methanol and Dimethyl Sulfoxide. Inorg. Chem. 2012, 51, 8827–8833. [Google Scholar] [CrossRef] [PubMed]
- Enderby, J.E. Ion Solvation via Neutron Scattering. Chem. Soc. Rev. 1995, 24, 159–168. [Google Scholar] [CrossRef]
- Badyal, Y.S.; Barnes, A.C.; Cuello, G.J.; Simonson, J.M. Understanding the effects of concentration on the solvation structure of Ca2+ in aqueous solutions. II: Insights into longer range order from neutron diffraction isotope substitution. J. Phys. Chem. A 2004, 108, 11819–11827. [Google Scholar] [CrossRef]
- Ansell, S.; Barnes, A.C.; Mason, P.E.; Neilson, G.W.; Ramos, S. X-ray and neutron scattering studies of the hydration structure of alkali ions in concentrated aqueous solutions. Biophys. Chem. 2006, 124, 171–179. [Google Scholar] [CrossRef]
- Mancinelli, R.; Botti, A.; Bruni, F.; Ricci, M.A.; Soper, A.K. Hydration of sodium, potassium, and chloride ions in solution and the concept of structure maker/breaker. J. Phys. Chem. B 2007, 111, 13570–13577. [Google Scholar] [CrossRef]
- Bruni, F.; Imberti, S.; Mancinelli, R.; Ricci, M.A. Aqueous solutions of divalent chlorides: Ions hydration shell and water structure. J. Chem. Phys. 2012, 136, 064520. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L.; McGreevy, R.L. MCGR: An inverse method for deriving the pair correlation function from the structure factor. Physica B 1997, 234–236, 357–358. [Google Scholar] [CrossRef]
- Pusztai, L. Partial pair correlation functions of liquid water. Phys. Rev. B 1999, 60, 11851–11854. [Google Scholar] [CrossRef]
- Soper, A.K. Tests of the empirical potential structure refinement method and a new method of application to neutron diffraction data on water. Mol. Phys. 2001, 99, 1503–1516. [Google Scholar] [CrossRef]
- Soper, A.K. Joint structure refinement of x-ray and neutron diffraction data on disordered materials: Application to liquid water. J. Phys. -Condens. Matter 2007, 19, 335206. [Google Scholar] [CrossRef]
- Harsanyi, I.; Pusztai, L. Hydration structure in concentrated aqueous lithium chloride solutions: A reverse Monte Carlo based combination of molecular dynamics simulations and diffraction data. J. Chem. Phys. 2012, 137, 204503–204509. [Google Scholar] [CrossRef]
- Mile, V.; Gereben, O.; Kohara, S.; Pusztai, L. On the Structure of Aqueous Cesium Fluoride and Cesium Iodide Solutions: Diffraction Experiments, Molecular Dynamics Simulations, and Reverse Monte Carlo Modeling. J. Phys. Chem. B 2012, 116, 9758–9767. [Google Scholar] [CrossRef]
- Pethes, I.; Pusztai, L. Reverse Monte Carlo modeling of liquid water with the explicit use of the SPC/E interatomic potential. J. Chem. Phys. 2017, 146, 064506. [Google Scholar] [CrossRef] [Green Version]
- Chialvo, A.A.; Simonson, J.M. The Structure of Concentrated NiCl2 Aqueous Solutions. What is Molecular Simulation Revealing about the Neutron Scattering Methodologies? Mol. Phys. 2002, 100, 2307–2315. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Simonson, J.M. The Structure of CaCl2 Aqueous Solutions over a Wide Range of Concentrations. Interpretation of Difraction Experiments Via Molecular Simulation. J. Chem. Phys. 2003, 119, 8052–8061. [Google Scholar] [CrossRef]
- Mason, P.E.; Neilson, G.W.; Dempsey, C.E.; Brady, J.W. Neutron diffraction and simulation studies of CsNO3 and Cs2CO3 solutions. J. Am. Chem. Soc. 2006, 128, 15136–15144. [Google Scholar] [CrossRef] [PubMed]
- Pluharova, E.; Fischer, H.E.; Mason, P.E.; Jungwirth, P. Hydration of the chloride ion in concentrated aqueous solutions using neutron scattering and molecular dynamics. Mol. Phys. 2014, 112, 1230–1240. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Vlcek, L. NO3– Coordination in Aqueous Solutions by 15N/14N and 18O/natO Isotopic Substitution: What Can We Learn from Molecular Simulation? J. Phys. Chem. B 2015, 119, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Kohagen, M.; Pluhařová, E.; Mason, P.E.; Jungwirth, P. Exploring Ion–Ion Interactions in Aqueous Solutions by a Combination of Molecular Dynamics and Neutron Scattering. J. Phys. Chem. Lett. 2015, 6, 1563–1567. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Vlcek, L. “Thought experiments” as dry-runs for “tough experiments”: Novel approaches to the hydration behavior of oxyanions. Pure Appl. Chem. 2016, 88, 163–176. [Google Scholar] [CrossRef]
- Jones, G.; Dole, M. The viscosity of aqueous solutions of strong electrolytes with special reference to barium chloride. J. Am. Chem. Soc. 1929, 51, 2950–2964. [Google Scholar] [CrossRef]
- Hepler, L.G. Thermal Expansion and Structure in Water and Aqueous Solutions. Can. J. Chem. 1969, 47, 4613–4617. [Google Scholar] [CrossRef]
- Falkenhagen, H.; Dole, M. The root law of the internal friction of strong electrolytes. Z. Fur Phys. Chem.-Abt. B-Chem. Der Elem. Aufbau Der Mater. 1929, 6, 159–162. [Google Scholar]
- Falkenhagen, H.; Dole, M. The internal friction of electrolytic solutions and their significance according to Debye’s theory. Phys. Z. 1929, 30, 611–622. [Google Scholar]
- Herskovits, T.T.; Kelly, T.M. Viscosity Studies of Aqueous Solutions of Alcohols, ureas, and Amides. J. Phys. Chem. 1973, 77, 381–388. [Google Scholar] [CrossRef]
- Rupley, J.A. Effect of Urea+amides upon the Water Structure. J. Phys. Chem. 1964, 68, 2002–2003. [Google Scholar] [CrossRef]
- Yoshida, K.; Tsuchihashi, N.; Ibuki, K.; Ueno, M. NMR and viscosity B coefficients for spherical nonelectrolytes in nonaqueous solvents. J. Mol. Liq. 2005, 119, 67–75. [Google Scholar] [CrossRef]
- Verissimo, L.M.P.; Cabral, I.; Cabral, A.M.T.D.P.V.; Utzeri, G.; Veiga, F.J.B.; Valente, A.J.M.; Ribeiro, A.C.F. Transport properties of aqueous solutions of the oncologic drug 5-fluorouracil: A fundamental complement to therapeutics. J. Chem. Thermodyn. 2021, 161, 106533. [Google Scholar] [CrossRef]
- Abbott, A.P.; Hope, E.G.; Palmer, D.J. Effect of solutes on the viscosity of supercritical solutions. J. Phys. Chem. B 2007, 111, 8114–8118. [Google Scholar] [CrossRef] [PubMed]
- Cox, W.M.; Wolfenden, J.H. The viscosity of strong electrolytes measured by a differential method. Proc. R. Soc. A. 1934, 145, 488. [Google Scholar]
- Bernal, J.D.; Fowler, R.H. A Theory of Water and Ionic Solution, with Particular Reference to Hydrogen and Hydroxyl Ions. J. Chem. Phys. 1933, 1, 515–548. [Google Scholar] [CrossRef]
- Laurence, V.D.; Wolfenden, J.H. The viscosity of solutions of strong electrolytes. J. Chem. Soc. 1934, 1144–1147. [Google Scholar] [CrossRef]
- Asmus, E. Zur Frage des B-Koeffizienten Der Jones-Dole Gleichung. Z. Fur Nat. Sect. A-A J. Phys. Sci. 1949, 4, 589–594. [Google Scholar] [CrossRef]
- Gurney, R.W. Ionic Processes in Solution; McGraw Hill: New York, NY, USA, 1953. [Google Scholar]
- Kaminsky, M. Ion-Solvent Interaction and the Viscosity of Strong Electrolyte Solutions. Discuss. Faraday Soc. 1957, 24, 171–179. [Google Scholar] [CrossRef]
- Nightingale, E.R. Phenomenological Theory of Ion Solvation: Effective Radii of Hydrated Ions. J. Phys. Chem. 1959, 63, 1381–1387. [Google Scholar] [CrossRef]
- Stokes, R.H.; Mills, R. Viscosity of Electrolytes and Related Properties; Pergamon Press: Oxford, UK; New York, NY, USA, 1965. [Google Scholar]
- Conway, B.E.; Verrall, R.E.; Desnoyer, J.E. Partial Molal Volumes of Tetraalkylamonium Halides and Assignment of Individual Ionic Contributions. Trans. Faraday Soc. 1966, 62, 2738–2749. [Google Scholar] [CrossRef]
- Frank, H.S.; Robinson, A.L. The Entropy of Dilution of Strong Electrolytes in Aqueous Solutions. J. Chem. Phys. 1940, 8, 933–938. [Google Scholar] [CrossRef]
- Frank, H.S.; Evans, M.W. Free Volume and Entropy in Condensed Systems. 3. Entropy in Binary Liquid Mixtures—Partial Molal Entropy in Dilute Solutions—Structure and Thermodynamics in Aqueous Electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Tsangaris, J.M.; Martin, R.B. Viscosities of Aqueous Solutions of Dipolar Ions. Arch. Biochem. Biophys. 1965, 112, 267–272. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Crisalle, O.D. Can Jones-Dole’s B-coefficient Be a Consistent Structure Making/Breaking Marker?. Rigorous molecular-based analysis and critical assessment of its marker uniqueness. J. Phys. Chem. B 2021, 125, 12028–12041. [Google Scholar] [CrossRef]
- Chialvo, A.A. On the Solute-Induced Structure-Making/Breaking Effect: Rigorous Links among Microscopic Behavior, Solvation Properties, and Solution Non-Ideality. J. Phys. Chem. B 2019, 123, 2930–2947. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Crisalle, O.D. Osmolyte-induced Effects on the Hydration Behavior and the Osmotic Second Virial Coefficients of Alkyl-Substituted Urea Derivatives. Critical assessment of their structure-making/breaking behavior. J. Phys. Chem. B 2021, 125, 6231–6243. [Google Scholar] [CrossRef]
- Kirkwood, J.G.; Buff, F.P. The Statistical Mechanical Theory of Solution. I. J. Chem. Phys. 1951, 19, 774–777. [Google Scholar] [CrossRef]
- Ball, P.; Hallsworth, J.E. Water structure and chaotropicity: Their uses, abuses and biological implications. Phys. Chem. Chem. Phys. 2015, 17, 8297–8305. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Crisalle, O.D. On density-based modeling of dilute non-electrolyte solutions involving wide ranges of state conditions and intermolecular asymmetries: Formal results, fundamental constraints, and the rationale for its molecular thermodynamic foundations. Fluid Phase Equilib. 2021, 535, 112969. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Cummings, P.T.; Kalyuzhnyi, Y.V. Solvation effect on kinetic rate constant of reactions in supercritical solvents. AlChE J. 1998, 44, 667–680. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Cummings, P.T. Comments on “Near Critical Phase Behavior of Dilute Mixtures”. Mol. Phys. 1995, 84, 41–48. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Crisalle, O.D. On the Linear Orthobaric-density Representation of Near-critical Solvation Quantities: What Can We Conclude about the Accuracy of this Paradigm? Fluid Phase Equilib. 2020, 514, 112535. [Google Scholar] [CrossRef]
- Chialvo, A.A. On the Krichevskii Parameter of Solutes in Dilute Solutions: Formal Links between its Magnitude, the Solute-solvent Intermolecular Asymmetry, and the Precise Description of Solution Thermodynamics. Fluid Phase Equilib. 2020, 513, 112546. [Google Scholar] [CrossRef]
- Levelt Sengers, J.M.H. Solubility Near the Solvent’s Critical Point. J. Supercrit. Fluids 1991, 4, 215–222. [Google Scholar] [CrossRef]
- Chialvo, A.A. Solute-Solute and Solute-Solvent Correlations in Dilute Near-Critical Ternary Mixtures: Mixed Solute and Entrainer Effects. J. Phys. Chem. 1993, 97, 2740–2744. [Google Scholar] [CrossRef]
- Brunner, G. (Ed.) Supercritical Fluids as Solvents and Reaction Media; Elsevier B.V.: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Anikeev, V.; Fan, M. Supercritical Fluid Technology for Energy and Environmental Applications; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Santos, D.T.; Santana, Á.L.; Meireles, M.A.A.; Gomes, M.T.M.S.; Torres, R.A.D.C.; Albarelli, J.Q.; Bakatselou, A.; Ensinas, A.V.; Maréchal, F. Supercritical Fluid Biorefining: Fundamentals, Applications and Perspectives; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Chialvo, A.A.; Crisalle, O.D. Solute-induced effects in solvation thermodynamics: Does urea behave as a structure-making or structure-breaking solute? Mol. Phys. 2019, 117, 3484–3492. [Google Scholar] [CrossRef]
- Chialvo, A.A. Molecular-Based Description of the Osmotic Second Virial Coefficients of Electrolytes: Rigorous Formal Links to Solute–Solvent Interaction Asymmetry, Virial Expansion Paths, and Experimental Evidence. J. Phys. Chem. B 2022, 126, 4339–4353. [Google Scholar] [CrossRef]
- Lin, L.N.; Brandts, J.F.; Brandts, J.M.; Plotnikov, V. Determination of the volumetric properties of proteins and other solutes using pressure perturbation calorimetry. Anal. Biochem. 2002, 302, 144–160. [Google Scholar] [CrossRef]
- Holtzer, A.; Emerson, M.F. On Utility of Concept of Water Structure in Rationalization of Aqueous Solutions of Proteins and Small Molecules. J. Phys. Chem. 1969, 73, 26–33. [Google Scholar] [CrossRef]
- Chialvo, A.A. Gas solubility in dilute solutions: A novel molecular thermodynamic perspective. J. Chem. Phys. 2018, 148, 174502. [Google Scholar] [CrossRef] [PubMed]
- Mazo, R.M. Statistical Mechanical Theory of Solutions. J. Chem. Phys. 1958, 29, 1122–1128. [Google Scholar] [CrossRef]
- Ben-Naim, A. Molecular Theory of Solutions; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Dole, M. Debye Contribution to the Theory of the Viscosity of Strong Electrolytes. J. Phys. Chem. 1984, 88, 6468–6469. [Google Scholar] [CrossRef]
- Falkenhagen, H.; Vernon, E.L. LXII. The viscosity of strong electrolyte solutions according to electrostatic theory. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1932, 14, 537–565. [Google Scholar] [CrossRef]
- Falkenhagen, H.; Hertz, H.G.; Eberling, W. Theorie der Elektrolyte; Hirzel: Leipzig, Germany, 1971. [Google Scholar]
- Robinson, R.A.; Stokes, R.H. Electrolyte Solutions, 2nd (revised) ed.; Butterworths: London, UK, 1970. [Google Scholar]
- Conway, B.E. Ionic Hydration in Chemistry and Biophysics; Elsevier: Amsterdam, The Netherlands, 1981; Volume 12. [Google Scholar]
- Franks, F. Water: A Matrix of Life; Royal Society of Chemistry: London, UK, 2000. [Google Scholar]
- Waghorne, W.E. Viscosities of electrolyte solutions. Philos. Trans. R. Soc. Lond. Ser. A-Math. Phys. Eng. Sci. 2001, 359, 1529–1543. [Google Scholar] [CrossRef]
- Hammadi, A. Electrical Conductance, Density, and Viscosity in Mixtures of Alkali-Metal Halides and Glycerol. Int. J. Thermophys. 2004, 25, 89–111. [Google Scholar] [CrossRef]
- Marcus, Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109, 1346–1370. [Google Scholar] [CrossRef]
- Dhondge, S.S.; Zodape, S.P.; Parwate, D.V. Volumetric and viscometric studies of some drugs in aqueous solutions at different temperatures. J. Chem. Thermodyn. 2012, 48, 207–212. [Google Scholar] [CrossRef]
- Jindal, R.; Singla, M.; Kumar, H. Transport behavior of aliphatic amino acids glycine/L-alanine/L-valine and hydroxyl amino acids L-serine/L-threonine in aqueous trilithium citrate solutions at different temperatures. J. Mol. Liq. 2015, 206, 343–349. [Google Scholar] [CrossRef]
- Gadzuric, S.; Tot, A.; Armakovic, S.; Armakovic, S.; Panic, J.; Jovic, B.; Vranes, M. Uncommon structure making/breaking behaviour of cholinium taurate in water. J. Chem. Thermodyn. 2017, 107, 58–64. [Google Scholar] [CrossRef]
- Zafarani-Moattar, M.T.; Shekaari, H.; Mostafavi, H.; Jafari, P. Thermodynamic and transport properties of aqueous solutions containing cholinium L-alaninate and polyethylene glycol dimethyl ether 250: Evaluation of solute-solvent interactions and phase separation. J. Chem. Thermodyn. 2019, 132, 9–22. [Google Scholar] [CrossRef]
- Sarma, T.S.; Ahluwalia, J.C. Experimental Studies on Structure of Aqueous Solutions of Hydrophobic Solutes. Chem. Soc. Rev. 1973, 2, 203–232. [Google Scholar] [CrossRef]
- Sanyal, S.K.; Mandal, S.K. Viscosity B-Coefficients of Alkyl Carboxylates. Electrochim. Acta 1983, 28, 1875–1876. [Google Scholar] [CrossRef]
- Tamaki, K.; Ohara, Y.; Watanabe, S. Solution Properties of Sodium Perfluoralkanoates. Heats of Solution, Viscosity B-coefficients, and Surface Tensions. Bull. Chem. Soc. Jpn. 1989, 62, 2497–2501. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H. Viscosity B-coefficients and standard partial molar volumes of amino acids, and their roles in interpreting the protein (enzyme) stabilization. Biophys. Chem. 2006, 122, 157–183. [Google Scholar] [CrossRef]
- Rajagopal, K.; Jayabalakrishnan, S.S. Volumetric, ultrasonic speed, and viscometric studies of salbutamol sulphate in aqueous methanol solution at different temperatures. J. Chem. Thermodyn. 2010, 42, 984–993. [Google Scholar] [CrossRef]
- Kabiraz, D.C.; Biswas, T.K.; Huque, M.E. Physico-chemical properties of some electrolytes in water and aqueous sodiumdodecyl sulfate solutions at different temperatures. J. Chem. Thermodyn. 2011, 43, 1917–1923. [Google Scholar] [CrossRef]
- Banipal, T.S.; Kaur, N.; Kaur, A.; Gupta, M.; Banipal, P.K. Effect of food preservatives on the hydration properties and taste behavior of amino acids: A volumetric and viscometric approach. Food Chem. 2015, 181, 339–346. [Google Scholar] [CrossRef]
- Kumar, K.; Patial, B.S.; Chauhan, S. Interactions of Saccharides in Aqueous Glycine and Leucine Solutions at Different Temperatures of (293.15 to 313.15) K: A Viscometric Study. J. Chem. Eng. Data 2015, 60, 47–56. [Google Scholar] [CrossRef]
- Sharma, S.K.; Singh, G.; Kumar, H.; Kataria, R. Densities, Sound Speed, and Viscosities of Some Amino Acids with Aqueous Tetra-Butyl Ammonium Iodide Solutions at Different Temperatures. J. Chem. Eng. Data 2015, 60, 2600–2611. [Google Scholar] [CrossRef]
- Banipal, T.S.; Beni, A.; Kaur, N.; Banipal, P.K. Volumetric, Viscometric and Spectroscopic Approach to Study the Solvation Behavior of Xanthine Drugs in Aqueous Solutions of NaCl at T = 288.15–318.15 K and at p = 101.325 kPa. J. Chem. Eng. Data 2017, 62, 20–34. [Google Scholar] [CrossRef]
- Zhu, C.Y.; Ren, X.F.; Ma, Y.G. Densities and Viscosities of Amino Acid plus Xylitol plus Water Solutions at 293.15 <= T/K <= 323.15. J. Chem. Eng. Data 2017, 62, 477–490. [Google Scholar] [CrossRef]
- Sharma, R.; Thakur, R.C.; Kumar, H. Study of viscometric properties of L-ascorbic acid in binary aqueous mixtures of D-glucose and D-fructose. Phys. Chem. Liq. 2019, 57, 139–150. [Google Scholar] [CrossRef]
- Sawhney, N.; Kumar, M.; Sandarve; Sharma, P.; Sharma, A.K.; Sharma, M. Structure-making behaviour of L-arginine in aqueous solution of drug ketorolac tromethamine: Volumetric, compressibility and viscometric studies. Phys. Chem. Liq. 2019, 57, 184–203. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, A.; Sharma, T.; Sharma, T.; Bamezai, R.K. Volumetric, acoustic and transport properties of ternary solutions of L-serine and L-arginine in aqueous solutions of thiamine hydrochloride at different temperatures. J. Chem. Thermodyn. 2019, 135, 260–277. [Google Scholar] [CrossRef]
- Sarmad, S.; Zafarani-Moattar, M.T.; Nikjoo, D.; Mikkola, J.P. How Different Electrolytes Can Influence the Aqueous Solution Behavior of 1-Ethyl-3-Methylimidazolium Chloride: A Volumetric, Viscometric, and Infrared Spectroscopy Approach. Front. Chem. 2020, 8, 593786. [Google Scholar] [CrossRef]
- Nain, A.K. Insight into solute-solute and solute-solvent interactions of l-proline in aqueous-D-xylose/L-arabinose solutions by using physicochemical methods at temperatures from 293.15 to 318.15 K. J. Mol. Liq. 2020, 318, 114190. [Google Scholar] [CrossRef]
- Rajput, P.; Richu; Sharma, T.; Kumar, A. Temperature dependent physicochemical investigations of some nucleic acid bases (uracil, thymine and adenine) in aqueous inositol solutions. J. Mol. Liq. 2021, 326, 115210. [Google Scholar] [CrossRef]
- Brinzei, M.; Ciocirlan, O. Volumetric and transport properties of ternary solutions of L-serine and L-valine in aqueous NaCl solutions in the temperature range (293.15–323.15) K. J. Chem. Thermodyn. 2021, 154, 106335. [Google Scholar] [CrossRef]
- Feakins, D.; Freemantle, D.J.; Lawrence, K.G. Transition-State Treatment of Relative Viscosity of Electrolyte Solutions: Applications to Aqueous, Non-Aqueous and Metanol+Water Systems. J. Chem. Soc.-Faraday Trans. I 1974, 70, 795–806. [Google Scholar] [CrossRef]
- Glasstone, S.; Laidler, K.J.; Eyring, H. The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena; McGraw-Hill Book Company, Incorporated: New York, NY, USA, 1941. [Google Scholar]
- Sedlbauer, J.; O’Connell, J.P.; Wood, R.H. A New Equation of State for Correlation and Prediction of Standard Molal Thermodynamic Properties of Aqueous Species at High Temperature and Pressures. Chem. Geol. 2000, 163, 43–63. [Google Scholar] [CrossRef]
- Sedlbauer, J.; Wood, R.H. Thermodynamic properties of dilute NaCl(aq) solutions near the critical point of water. J. Phys. Chem. B 2004, 108, 11838–11849. [Google Scholar] [CrossRef]
- Panic, J.; Vranes, M.; Tot, A.; Ostojic, S.; Gadzuric, S. The organisation of water around creatine and creatinine molecules. J. Chem. Thermodyn. 2019, 128, 103–109. [Google Scholar] [CrossRef]
- Richu; Kumar, A. A Comprehensive Study on Molecular Interactions of l-Ascorbic Acid/Nicotinic Acid in Aqueous [BMIm]Br at Varying Temperatures and Compositions: Spectroscopic and Thermodynamic Insights. J. Chem. Eng. Data 2021, 66, 3859–3880. [Google Scholar] [CrossRef]
- Hossain, M.F.; Biswas, T.K.; Islam, M.N.; Huque, M.E. Volumetric and viscometric studies on dodecyltrimethylammonium bromide in aqueous and in aqueous amino acid solutions in premicellar region. Monatsh. Chem. 2010, 141, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Vranes, M.; Tot, A.; Jankovic, N.; Gadzuric, S. What is the taste of vitamin-based ionic liquids? J. Mol. Liq. 2019, 276, 902–909. [Google Scholar] [CrossRef]
- Rudan-Tasic, D.; Klofutar, C.; Horvat, J. Viscosity of aqueous solutions of some alkali cyclohexylsulfamates at 25.0 degrees C. Food Chem. 2004, 86, 161–167. [Google Scholar] [CrossRef]
- Horvat, J.; Bester-Rogac, M.; Klofutar, C.; Rudan-Tasic, D. Viscosity of aqueous solutions of lithium, sodium, potassium, rubidium and caesium cyclohexylsulfamates from 293.15 to 323.15 k. J. Solut. Chem. 2008, 37, 1329–1342. [Google Scholar] [CrossRef]
- Dunn, L.A. Apparent Molar Volumes of Electrolytes. 3: Some 1-1 and 2-1 Electrolytes in Aqueous Solution at 0, 5. 15. 35, 45, 55, and 65 Degrees C. Trans. Faraday Soc. 1968, 64, 2951–2961. [Google Scholar] [CrossRef]
- Abdulagatov, I.M.; Azizov, N.D. Viscosity of aqueous calcium chloride solutions at high temperatures and high pressures. Fluid Phase Equilib. 2006, 240, 204–219. [Google Scholar] [CrossRef]
- Herrington, T.M.; Roffey, M.G.; Smith, D.P. Densities of Aqueous Electrolytes MnCl2, CoCl2, NiCl2, ZnCl2, and CdCl2 from 25 degrees to 72 degrees at 1atm. J. Chem. Eng. Data 1986, 31, 221–225. [Google Scholar] [CrossRef]
- Sonika; Thakur, R.C.; Chauhan, S.; Singh, K. Molecular Interactions of Transition Metal Chlorides in Water and Water–Ethanol Mixtures at 298–318 K on Viscometric Data. Russ. J. Phys. Chem. A 2018, 92, 2701–2709. [Google Scholar] [CrossRef]
- Apelblat, A. A new two-parameter equation for correlation and prediction of densities as a function of concentration and temperature in binary aqueous solutions. J. Mol. Liq. 2016, 219, 313–331. [Google Scholar] [CrossRef]
- Clegg, S.L.; Wexler, A.S. Densities and Apparent Molar Volumes of Atmospherically Important Electrolyte Solutions. 1. The Solutes H2SO4, HNO3, HCl, Na2SO4, NaNO3, NaCl, (NH4)2SO4, NH4NO3, and NH4Cl from 0 to 50 degrees C, Including Extrapolations to Very Low Temperature and to the Pure Liquid State, and NaHSO4, NaOH, and NH3 at 25 degrees C. J. Phys. Chem. A 2011, 115, 3393–3460. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.N.; Friesen, R.J. Conductance in the Range of Medium Concentration. Can. J. Chem.-Rev. Can. De Chim. 1959, 37, 1288–1293. [Google Scholar] [CrossRef]
- Laliberte, M.; Cooper, W.E. Model for calculating the density of aqueous electrolyte solutions. J. Chem. Eng. Data 2004, 49, 1141–1151. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Cummings, P.T. Solute-induced Effects on the Structure and the Thermodynamics of Infinitely Dilute Mixtures. AlChE J. 1994, 40, 1558–1573. [Google Scholar] [CrossRef]
- Kim, S.; Johnston, K.P. Clustering in Supercritical Fluid Mixtures. AlChE J. 1987, 33, 1603–1611. [Google Scholar] [CrossRef]
- Wu, R.-S.; Lee, L.L.; Cochran, H.D. Solvent Structural Changes in Repulsive and Attractive Supercritical Mixtures. A Molecular Distribution Study. J. Supercrit. Fluids 1992, 5, 192–198. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Tomasko, D.L.; Peshkin, J.; Eckert, C.A. Fluorescence Spectroscopy Studies of Dilute Supercritical Solutions. Ind. Eng. Chem. Res. 1990, 29, 1682–1690. [Google Scholar] [CrossRef]
- Eckert, C.A.; Knutson, B.L. Molecular Charisma in Supercritical Fluids. Fluid Phase Equilib. 1993, 83, 93–100. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Debenedetti, P.G.; Eckert, C.A.; Johnston, K.P. Letter to the Editor. AlChE J. 1990, 36, 1927–1928. [Google Scholar] [CrossRef]
- Economou, I.G.; Donohue, M.D. Mean Field Calculation of Thermodynamic Properties of Supercritical Fluids. AlChE J. 1990, 36, 1920–1925. [Google Scholar] [CrossRef]
- Feynman, R.P. What Do You Care What Other People Think?: Further Adventures of a Curious Character; Norton & Company, Incorporated: New York, NY, USA, 1988. [Google Scholar]
- Vraneš, M.; Rackov, S.; Papović, S.; Pilić, B. The study of interactions in aqueous solutions of 1-alkyl-3-(3-butenyl)imidazolium bromide ionic liquids. J. Chem. Thermodyn. 2021, 159, 106479. [Google Scholar] [CrossRef]
- Gupta, J.; Chand, D.; Nain, A.K. Study to reconnoiter solvation consequences of l-arginine/l-histidine and sodium salicylate in aqueous environment probed by physicochemical approach in the temperature range (293.15–318.15) K. J. Mol. Liq. 2020, 305, 112848. [Google Scholar] [CrossRef]
- Ankita; Nain, A.K. Solute-solute and solute-solvent interactions of drug sodium salicylate in aqueous-glucose/sucrose solutions at temperatures from 293.15 to 318.15 K: A physicochemical study. J. Mol. Liq. 2020, 298, 112006. [Google Scholar] [CrossRef]
- Majer, V.; Sedlbauer, J.; Wood, R.H. Chapter 4—Calculation of standard thermodynamic properties of aqueous electrolytes and nonelectrolytes. In Aqueous Systems at Elevated Temperatures and Pressures; Palmer, D.A., Fernández-Prini, R., Harvey, A.H., Eds.; Academic Press: London, UK, 2004; pp. 99–147. [Google Scholar] [CrossRef]
- Sedlbauer, J.; (Department of Chemistry, Technical University of Liberec, Liberec, Czechia). Personal communication, 2022.



{kind=link}
{kind=link}
| Ref. | |||||
|---|---|---|---|---|---|
| Water (LR-IS) | 0 | 0 | 0 | maker | This work |
| Ideal gas | maker | <0 | <0 | breaker | This work |
| creatine | breaker | <0 | >0 | maker | [105] |
| creatinine | breaker | <0 | >0 | maker | [105] |
| nicotinic acid | breaker | <0 | >0 | maker | [106] |
| l-ascorbic acid | breaker | >0 | >0 | breaker | [50,94,106] |
| glycine | breaker | >0 | >0 | breaker | [107] |
| alanine | breaker | >0 | >0 | breaker | [107] |
| DTAB (c) | breaker | >0 | <0 | breaker | [107] |
| l-serine | breaker | <0 | >0 | maker | [96] |
| l-arginine | breaker | <0 | >0 | maker | [96] |
| choline-biotinate | breaker | <0 | >0 | maker | [108] |
| choline-nicotinate | breaker | <0 | >0 | maker | [108] |
| choline-ascorbate | breaker | >0 | >0 | maker | [108] |
| LiCy (d) | breaker | <0 | >0 | breaker | [109,110] |
| NaCy (d) | breaker | ~0 | >0 | maker | [109,110] |
| KCy (d) | breaker | >0 | >0 | maker | [109,110] |
| CaCl2 | maker | >0 | >0 | breaker | [111,112] |
| CdCl2 | maker | <0 | >0 | breaker | [113,114] |
| NiCl2 | maker | >0 | <0 | breaker (e) | [114,115] |
| NH4NO3 | breaker | >0 | <0 | breaker | [116,117] |
| MgCl2 | maker | <0 | >0 | breaker | [114,118] |
| CaCl2 | >0 | >0 | >0 (maker) |
| choline-ascorbate | >0 | >0 | <0 (breaker) |
| CdCl2 | >0 | <0 | >0 (maker) |
| NH4NO3 | >0 | <0 | <0 (breaker) |
| MgCl2 | <0 | >0 | >0 (maker) |
| LiCy (a) | <0 | >0 | <0 (breaker) |
| IG_β | <0 | <0 | >0 (maker) |
| IG_β (b) | <0 | <0 | <0 (breaker) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chialvo, A.A.; Crisalle, O.D. Solute-Induced Perturbation of the Solvent Microstructure in Aqueous Electrolyte Solutions: Some Uses and Misuses of Structure Making/Breaking Criteria. Liquids 2022, 2, 106-130. https://doi.org/10.3390/liquids2030008
Chialvo AA, Crisalle OD. Solute-Induced Perturbation of the Solvent Microstructure in Aqueous Electrolyte Solutions: Some Uses and Misuses of Structure Making/Breaking Criteria. Liquids. 2022; 2(3):106-130. https://doi.org/10.3390/liquids2030008
Chicago/Turabian StyleChialvo, Ariel A., and Oscar D. Crisalle. 2022. "Solute-Induced Perturbation of the Solvent Microstructure in Aqueous Electrolyte Solutions: Some Uses and Misuses of Structure Making/Breaking Criteria" Liquids 2, no. 3: 106-130. https://doi.org/10.3390/liquids2030008