
Hydrolysis of Al3+ in Aqueous Solutions: Experiments and Ab Initio Simulations

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.2. Potentiometric Equipment and Procedure
2.3. Calculation Programs for Equilibria in Solution
2.4. Quantum-Mechanical Calculations and Ab Initio Molecular Dynamics Simulations
3. Results and Discussion
3.1. Ionic Strength Dependence and Species with Chloride
3.2. Quantum-Based Simulations of Al Complexes in Implicit and Explicit Aqueous Solvents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yokel, R.A. Aluminum. In Elements and their Compounds in the Environment–Occurrence, Analysis and Biological Relevance, 2nd completely and enlarged edition ed.; Merian, E., Anke, M., Ihnat, M., Stoeppler, M., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Volume 2–Metals and their compounds; pp. 635–658. [Google Scholar]
- Kiss, T. From coordination chemistry to biological chemistry of aluminium. J. Inorg. Biochem. 2013, 128, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.R. Aluminum speciation. In Encyclopedia of Soils in the Environment; Hillel, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 50–56. [Google Scholar]
- Yokel, R.A.; McNamara, P.J. Aluminum toxicocinetics: An updated mini-review. Pharmacol. Toxicol. 2001, 88, 159–167. [Google Scholar] [CrossRef]
- Baes, C.F.; Mesmer, R.E. The Hydrolysis of Cations; John Wiley & Sons: New York, NY, USA, 1976. [Google Scholar]
- Salvatore, F.; Trifuoggi, M. Investigations of Polyoxometalates in Aqueous Solutions. I. The Formation of Al13(OH)327+ Cation. J. Coord. Chem. 2000, 51, 271–282. [Google Scholar] [CrossRef]
- Brown, P.L.; Sylva, R.N.; Batley, G.E.; Ellis, J. The hydrolysis of metal ions. Part 8. Aluminium(III). J. Chem. Soc. Dalton Trans. 1985, 9, 1967–1970. [Google Scholar] [CrossRef]
- Cigala, R.M.; De Stefano, C.; Giacalone, A.; Gianguzza, A. Speciation of Al3+ in fairly concentrated solutions (20–200 mmol L−1) at I = 1 mol L−1 (NaNO3), in the acidic pH range, at different temperatures. Chem. Spec. Bioavail. 2011, 23, 33–37. [Google Scholar] [CrossRef]
- De Stefano, C.; Sammartano, S.; Mineo, P.; Rigano, C. Computer Tools for the Speciation of Natural Fluids. In Marine Chemistry–An Environmental Analytical Chemistry Approach; Gianguzza, A., Pelizzetti, E., Sammartano, S., Eds.; Kluwer Academic Publishers: Amsterdam, The Netherlands, 1997; pp. 71–83. [Google Scholar]
- Gumienna-Kontecka, E.; Berthon, G.; Fritsky, I.O.; Wieczorek, R.; Latajka, Z.; Kozlowski, H. 2-(Hydroxyimino)propanohydroxamic acid, a new effective ligand for aluminium. J. Chem. Soc. Dalton Trans. 2000, 9, 4201–4208. [Google Scholar] [CrossRef]
- Martell, A.E.; Smith, R.M.; Motekaitis, R.J. Critically Selected Stability Constants of Metal Complexes. Gaithersburg; National Institute of Standard and Technology, NIST: Gaithersburg, MD, USA, 2004. [Google Scholar]
- May, P.M.; Murray, K. Database of chemical reactions designed to achieve thermodynamic consistency automatically. J. Chem. Eng. Data 2001, 46, 1035–1040. [Google Scholar] [CrossRef]
- Pettit, L.D.; Powell, K.J. IUPAC Stability Constants Database; Academic Software, IUPAC: Research Triangle Park, NC, USA, 2001. [Google Scholar]
- Sarpola, A.; Hellman, H.; Hietapelto, V.; Jalonen, J.; Jokela, J.; Ramo, J.; Saukkoriipi, J. Hydrolysis products of water treatment chemical aluminium sulfate octadecahydrate by electrospray ionization mass spectrometry. Polyhedron 2007, 26, 2851–2858. [Google Scholar] [CrossRef]
- Gao, B.; Yue, Q. Effect of SO42−/Al3+ ratio and OH−/Al3+ value on the characterization of coagulant poly-aluminum-chloride-sulfate (PACS) and its coagulation performance in water treatment. Chemosphere 2005, 61, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Shi, W.; Zhang, J.; Bi, S. 27Al NMR Chemical Shifts and Relative Stabilities of Aqueous Monomeric Al3+ Hydrolytic Species with Different Coordination Structures. ACS Earth Space Chem. 2019, 3, 1353–1361. [Google Scholar] [CrossRef]
- Li, C.; Liu, W.; Ma, Y. Influence of H3O+ on the structure formation of oligomers in aluminium sols prepared from basic aluminium acetate: Experiments and computations. J. Mol. Liq. 2019, 289, 111052. [Google Scholar] [CrossRef]
- Maki, H.; Sakatab, G.; Mizuhata, M. Quantitative NMR of quadrupolar nucleus as a novel analytical method: Hydrolysis behaviour analysis of aluminum ion. Analyst 2017, 142, 1790–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nýblová, D.; Senna, M.; Düvel, A.; Heitjans, P.; Billik, P.; Filo, J.; Šepelák, V. NMR study on reaction processes from aluminum chloride hydroxides to alpha alumina powders. J. Am. Ceram Soc. 2019, 102, 2871–2881. [Google Scholar]
- Bertsch, P.M.; Thomas, G.W.; Barnhisel, R.I. Characterization of hydroxy-aluminum solutions by aluminum-27 nuclear magnetic resonance spectroscopy. Soil Sci. Soc. Am. J. 1986, 50, 825–830. [Google Scholar] [CrossRef]
- Buffle, J.; Parthasarathy, N.; Haerdi, W. Importance of speciation methods in analytical control of water treatment processes with application to fluoride removal from waste waters. Water Res. 1985, 19, 7–23. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, B.; Lee, C. Effect of low temperature on aluminum(III) hydrolysis: Theroretical and experimental studies. J. Environ. Sci. 2008, 20, 907–914. [Google Scholar]
- Yang, X. Spectroscopy study of aluminium speciation in removing humic substances by Al coagulation. Water Res. 1999, 33, 3271–3280. [Google Scholar]
- Li, N.; Hu, C.; Fu, X.; Xu, X.; Liu, R.; Liu, H.; Qu, J. Identification of Al13 on the colloid surface using Surface-Enhanced Raman Spectroscopy. Environ. Sci. Technol. 2017, 51, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Suarez, D.L. In situ infrared speciation of adsorbed carbonate on aluminum and iron oxides. Clays Clay Miner. 1997, 45, 814–825. [Google Scholar] [CrossRef]
- Sipos, P.; Capewell, S.G.; May, P.M.; Hefter, G.; Laurenczy, G.; Lukács, F.; Roulet, R. Spectroscopic studies of the chemical speciation in concentrated alkaline aluminate solutions. J. Chem. Soc. Dalton Trans. 1998, 18, 3007–3012. [Google Scholar] [CrossRef]
- Marin, C.; Tudorache, A.; Vladescu, L. Aluminium Determination and Speciation Modelling in Groundwater from the Area of a Future Radioactive Waste Repository. Rev. Chim. 2010, 61, 431–438. [Google Scholar]
- Zhao, H.; Hu, C.Z.; Liu, H.J.; Zhao, X.; Qu, J.H. Role of aluminum speciation in the removal of disinfection by product precursors by a coagulation process. Environ. Sci. Technol. 2008, 42, 5752–5758. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, W.; Ma, Y. DFT Studies on the Al-Speciation and Its Structure in Aqueous Aluminum Sol Formed by Aluminum Formoacetate. J. Phys. Chem. B 2019, 123, 9167–9179. [Google Scholar] [CrossRef]
- Dong, S.; Shi, W.; Zhang, J.; Bi, S. DFT Studies on the Water-Assisted Synergistic Proton Dissociation Mechanism for the Spontaneous Hydrolysis Reaction of Al3+ in Aqueous Solution. ACS Earth Space Sci. 2018, 2, 269–277. [Google Scholar] [CrossRef]
- Jin, X.; Yang, W.; Qian, Z.; Wanga, Y.; Bi, S. DFT study on the interaction between monomeric aluminium and chloride ion in aqueous solution. Dalton Trans. 2011, 40, 5052–5058. [Google Scholar] [CrossRef] [PubMed]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frish, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Frish, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Frish, M.J.; Pople, J.A. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Model. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schutt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package–Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Previti, E.; Foti, C.; Giuffrè, O.; Saija, F.; Sponer, J.; Cassone, G. Ab initio molecular dynamics simulations and experimental speciation study of levofloxacin under different pH conditions. Phys. Chem. Chem. Phys. 2021, 23, 24403–24412. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G. Nuclear Quantum Effects largely influence molecular dissociation and proton transfer in liquid water under an electric field. J. Phys. Chem. Lett. 2020, 11, 8983–8988. [Google Scholar] [CrossRef]
- Cassone, G.; Creazzo, F.; Giaquinta, P.V.; Saija, F.; Saitta, A.M. Ab initio molecular dynamics study of an aqueous NaCl solution under an electric field. Phys. Chem. Chem. Phys. 2016, 18, 23164–23173. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 26, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filella, M.; May, P.M. Reflections on the calculation and publication of potentiometrically-determined formation constants. Talanta 2005, 65, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Cardiano, P.; De Stefano, C.; Giuffrè, O.; Sammartano, S. Thermodynamic and spectroscopic study for the interaction of dimethyltin(IV) with L-cysteine in aqueous solution. Biophys. Chem. 2008, 133, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, G.; Giuffrè, O.; Sammartano, S. Acid-base and UV properties of some aminophenol ligands and their complexing ability towards Zn2+ in aqueous solution. J. Mol. Liquids 2011, 159, 146–151. [Google Scholar] [CrossRef]
- Abate, C.; Cassone, G.; Cordaro, M.; Giuffrè, O.; Mollica-Nardo, V.; Ponterio, R.C.; Saija, F.; Sponer, J.; Trusso, S.; Foti, C. Understanding the behaviour of carnosine in aqueous solution: An experimental and quantum-based computational investigation on acid-base properties and complexation mechanisms with Ca2+ and Mg2+. New J. Chem. 2021, 45, 20352–20364. [Google Scholar] [CrossRef]
- Cassone, G.; Chillè, D.; Foti, C.; Giuffrè, O.; Ponterio, R.C.; Sponer, J.; Saija, F. Stability of hydrolytic arsenic species in aqueous solutions: As3+ vs. As5+. Phys. Chem. Chem. Phys. 2018, 20, 23272–23280. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G.; Chillè, D.; Mollica Nardo, V.; Giuffrè, O.; Ponterio, R.C.; Sponer, J.; Trusso, S.; Saija, F.; Foti, C. Arsenic-nucleotides interactions: An experimental and computational investigation. Dalton Trans. 2020, 49, 6302–6311. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
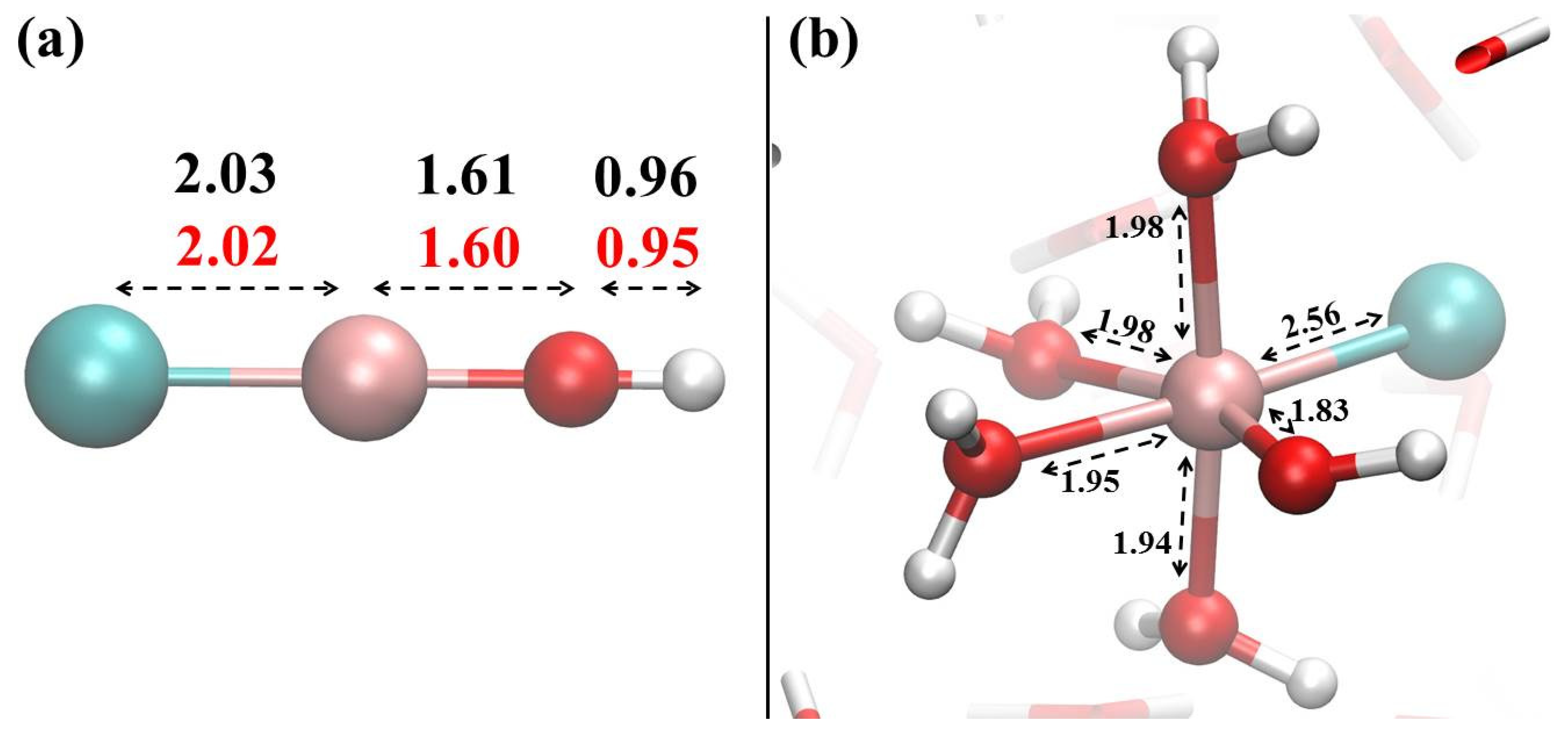{kind=link}
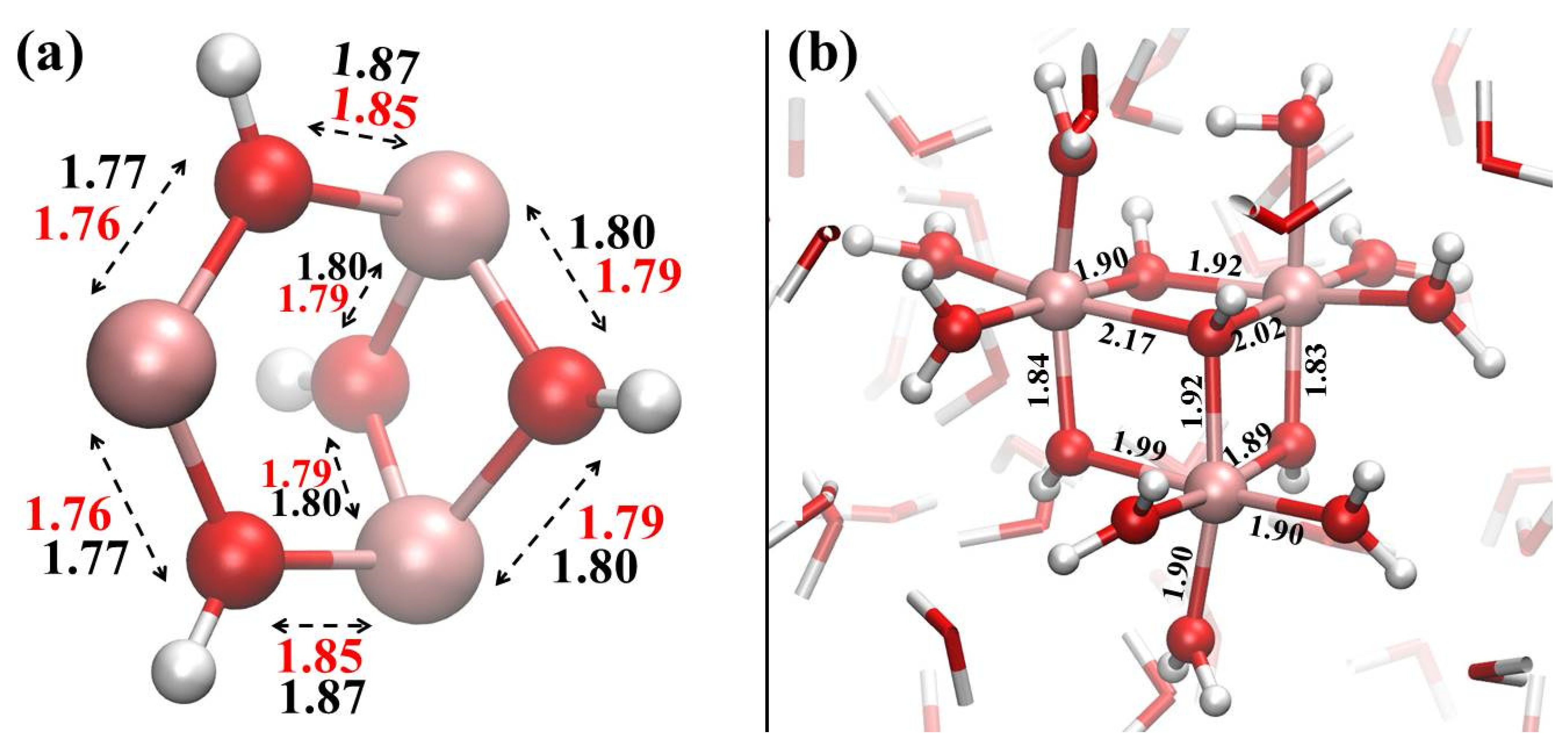{kind=link}
| CAl /mmol L−1 | Ionic Medium | CCl/mol L−1 | CNO3/mol L−1 | CNa/mol L−1 |
|---|---|---|---|---|
| 10–20 | NaCl | 0.1 | — | 0.1 |
| 10–20 | 1 | — | 1 | |
| 10–20 | NaNO3 | — | 0.1 | 0.1 |
| 10–20 | — | 1 | 1 | |
| NaCl/NaNO3 | ||||
| 5–20 | 0.25 | 0.25 | 0.5 | |
| 5–20 | 0.25 | 0.75 | 1 | |
| 5–20 | 0.75 | 0.25 | 1 | |
| 20 | 0.5 | 0.5 | 1 |
| Ionic Medium | I1 | CAl 1 | CCl 1 | CNO3 1 | logβpq 2 | |
|---|---|---|---|---|---|---|
| Al3(OH)45+ | Al13(OH)327+ | |||||
| NaCl | 0.142 | 0.015 | 0.11 | — | −13.324(3) 3 | −108.48(2) 3 |
| 0.946 | 0.015 | 1.02 | — | −13.618(3) | −112.72(1) | |
| NaNO3 | 0.167 | 0.015 | — | 0.16 | −13.272(3) | −108.38(1) |
| 0.963 | 0.015 | — | 1.03 | −13.320(3) | −110.43(1) | |
| NaCl/NaNO3 | 0.505 | 0.012 | 0.26 | 0.28 | −13.248(4) | −109.88(2) |
| 0.939 | 0.012 | 0.26 | 0.76 | −13.241(5) | −110.62(1) | |
| 0.949 | 0.012 | 0.75 | 0.27 | −13.314(3) | −111.30(1) | |
| 0.919 | 0.02 | 0.50 | 0.55 | −13.304(7) | −110.97(2) |
| Species | Ionic Medium | logTβpq 1 | C |
|---|---|---|---|
| Al3(OH)45+ | NaCl | −13.490(1) 2 | −0.562(6) 2 |
| Al13(OH)327+ | −103.81(6) | −1.75(7) | |
| Al3(OH)45+ | NaNO3 | −13.490(1) | −0.244(6) |
| Al13(OH)327+ | −103.81(6) | 0.69(8) | |
| Al3(OH)45+ | NaCl/NaNO3 | −13.490(1) | −0.219(2) |
| Al13(OH)327+ | −103.81(6) | 0.1(1) |
| Species | logTβ | C1 |
|---|---|---|
| Al3(OH)45+ 2 | −13.44(2) 3 | −0.23(2) 3 |
| Al13(OH)327+ 2 | −103.89(4) | 1.43(8) |
| AlCl2+ 4 | 1.33(3) | −0.32(5) |
| AlClOH+ 5 | −3.26(5) | 0.26(7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacobello, F.; Mollica-Nardo, V.; Foti, C.; Ponterio, R.C.; Saija, F.; Trusso, S.; Sponer, J.; Cassone, G.; Giuffrè, O. Hydrolysis of Al3+ in Aqueous Solutions: Experiments and Ab Initio Simulations. Liquids 2022, 2, 26-38. https://doi.org/10.3390/liquids2010003
Giacobello F, Mollica-Nardo V, Foti C, Ponterio RC, Saija F, Trusso S, Sponer J, Cassone G, Giuffrè O. Hydrolysis of Al3+ in Aqueous Solutions: Experiments and Ab Initio Simulations. Liquids. 2022; 2(1):26-38. https://doi.org/10.3390/liquids2010003
Chicago/Turabian StyleGiacobello, Fausta, Viviana Mollica-Nardo, Claudia Foti, Rosina Celeste Ponterio, Franz Saija, Sebastiano Trusso, Jiri Sponer, Giuseppe Cassone, and Ottavia Giuffrè. 2022. "Hydrolysis of Al3+ in Aqueous Solutions: Experiments and Ab Initio Simulations" Liquids 2, no. 1: 26-38. https://doi.org/10.3390/liquids2010003