
Porphyromonas gingivalis Strain W83 Infection Induces Liver Injury in Experimental Alcohol-Associated Liver Disease (ALD) in Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals Fed with Alcohol
2.2. Bacteria Preparation and Mouse Treatment
2.3. RNA Isolation and Real-Time Polymerase Chain Reaction (Real-Time PCR)
2.4. Western Blot Analysis, Serum Lcn2 ELISA Assay and Serum AST/ALT Activity Measurement
2.5. Immunohistochemistry Staining
2.6. Liver Tissue Hematoxylin and Eosin Staining (H&E Staining) [21]
2.7. Naphthol AS-D Chloroacetate Esterase (CAE) Staining
2.8. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
2.9. Statistical Analysis
3. Results
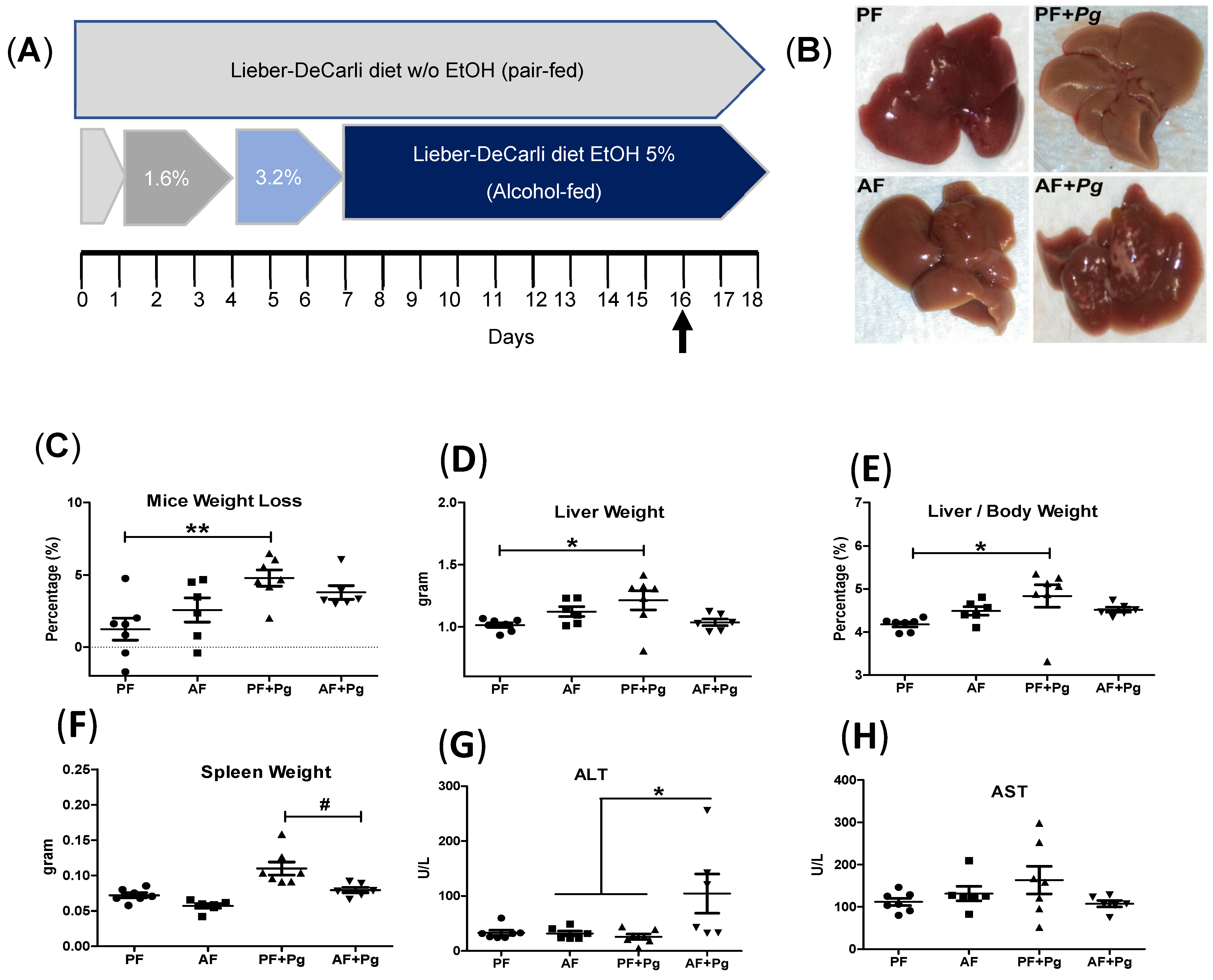
3.1. Pg Infection of Alcohol-Fed Mice Induced Liver Injury
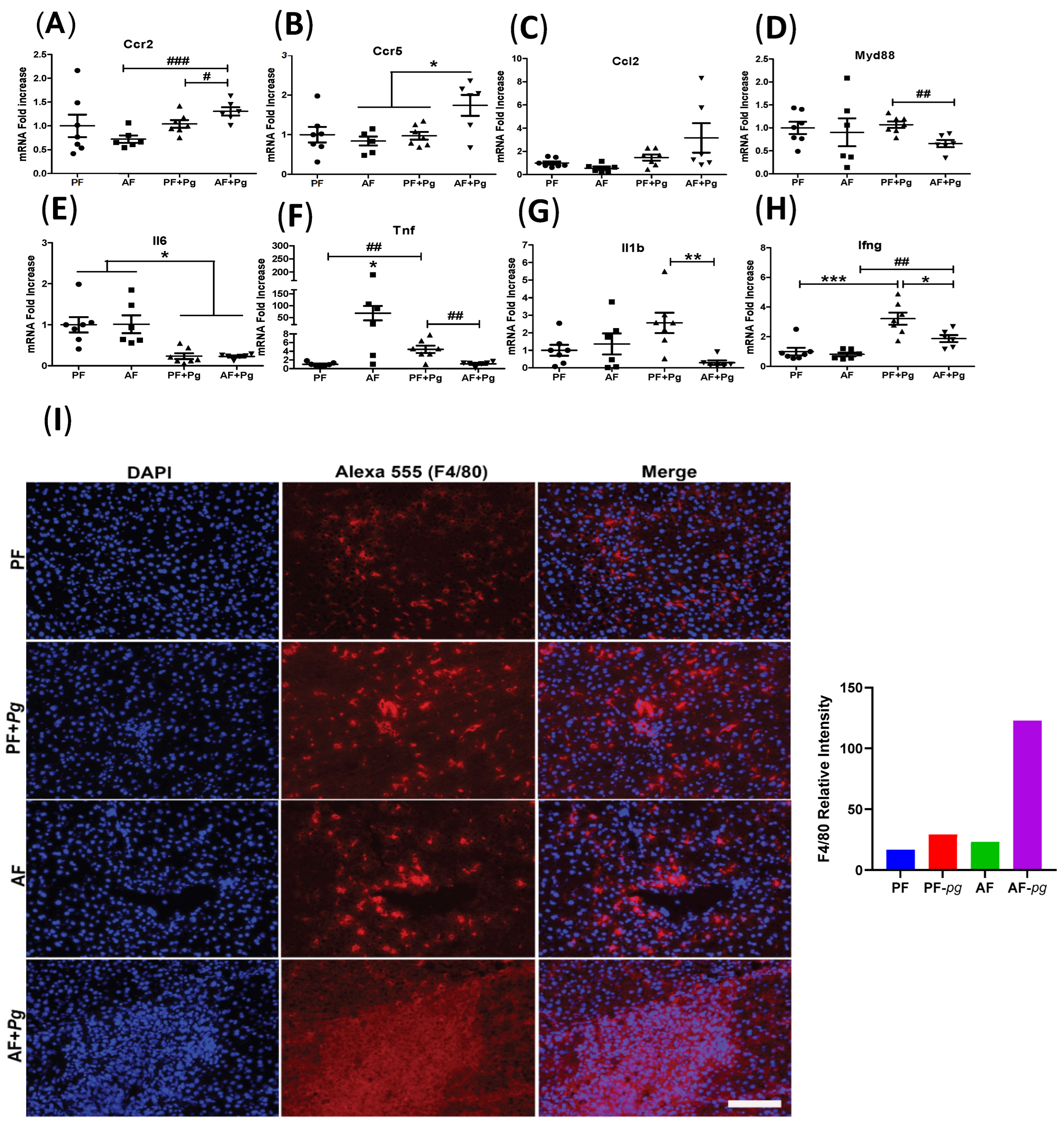
3.2. Pg Infection of Alcohol-Fed Mice Induced Infiltration of Inflammatory Monocytes/Macrophages but Repressed Expression of Inflammatory Cytokines
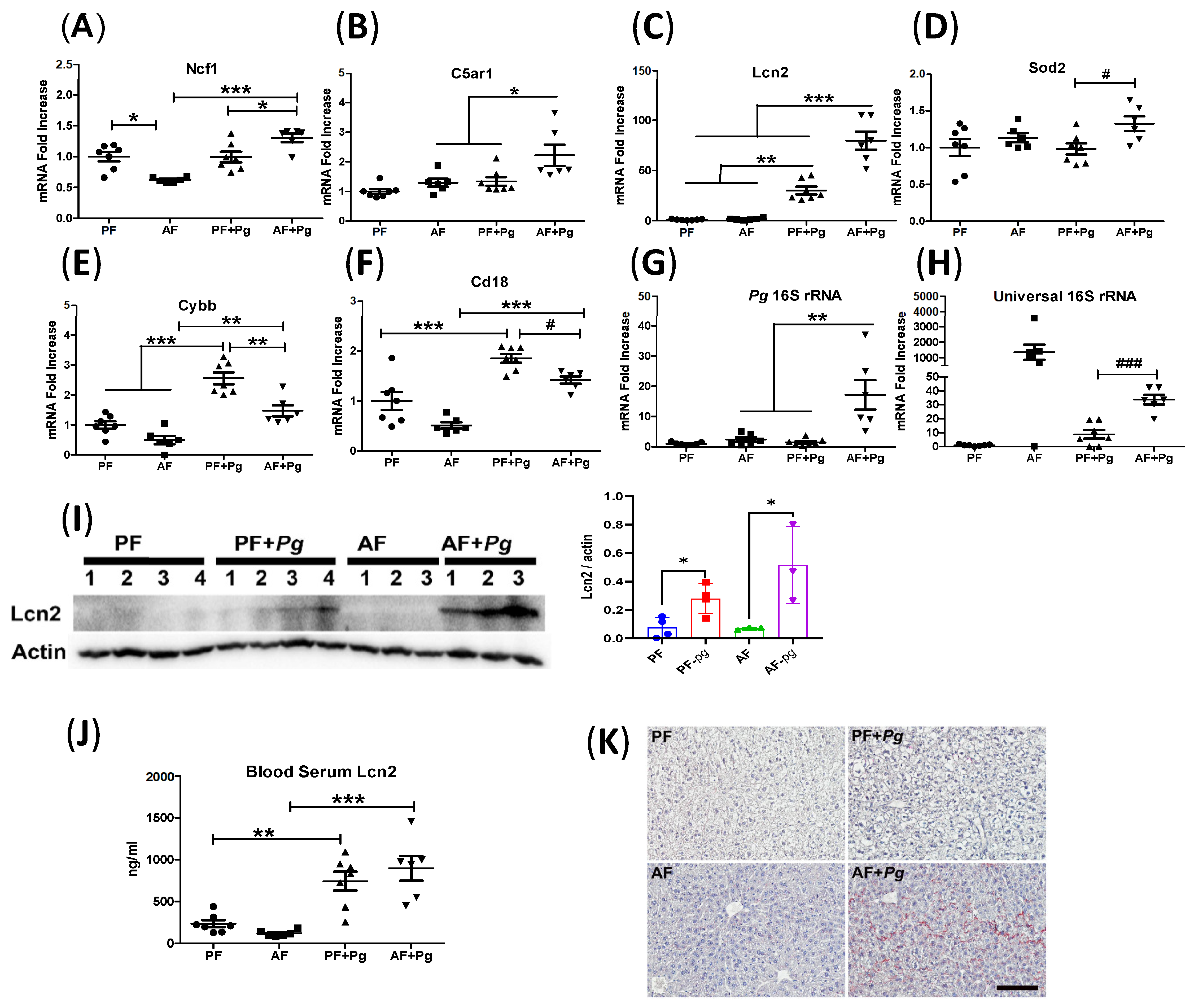
3.3. Pg Infection of Alcohol-Fed Mice Induced Neutrophil Infiltration and Defective Bacterial Clearance
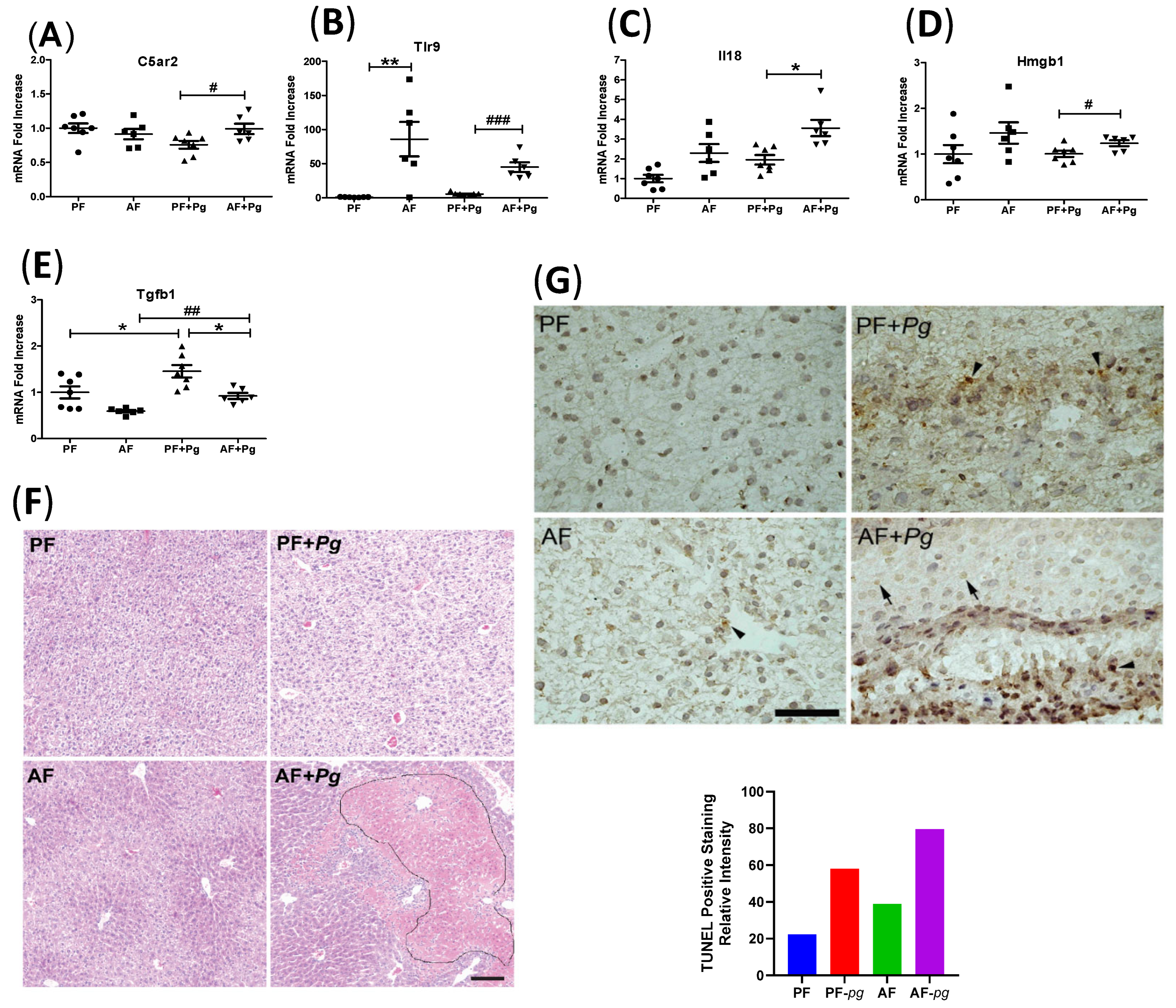
3.4. Pg Infection of Alcohol-Fed Mice Induced Inflammasome Activation and Cell Death
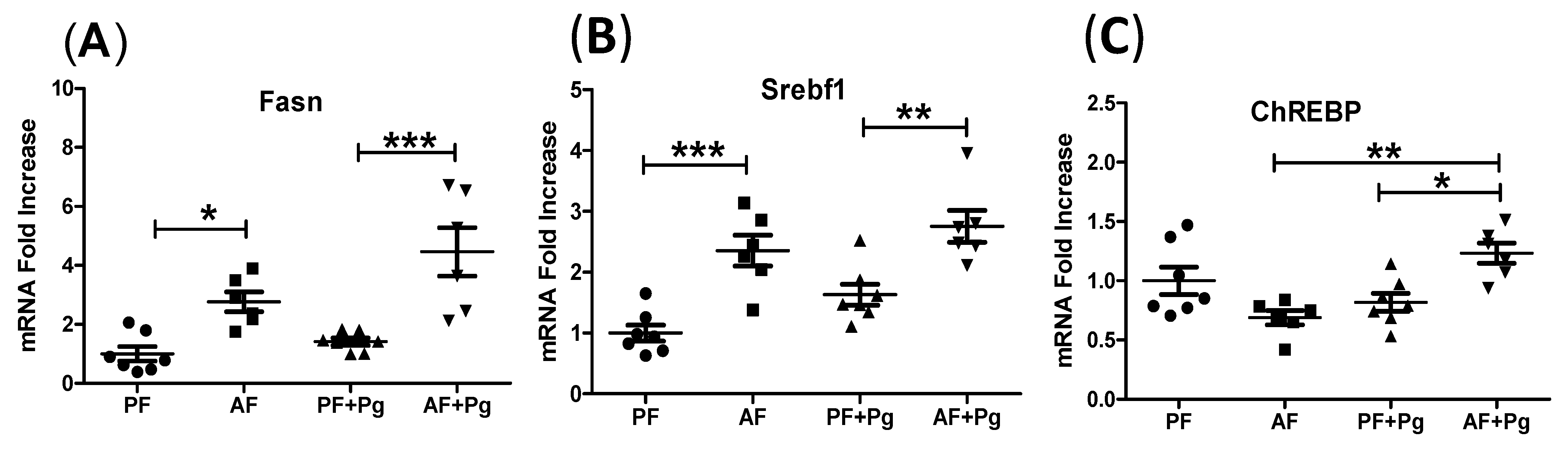
3.5. Pg Infection of Alcohol-Fed Mice Induced Lipogenesis-Related Gene Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AH | alcohol-associated steatohepatitis |
| AF | alcohol-fed |
| ALD | alcohol-associated liver diseases |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| C5ar | C5a receptor |
| Cr3 | complement receptor 3 |
| CFU | colony-forming units |
| H and E | hematoxylin and eosin |
| Hmgb1 | high mobility group box-1 protein gene |
| IP | intraperitoneal injection |
| KC | Kupffer cell |
| Lcn2 | lipocalin 2 |
| Ncf1 | Neutrophil cytosol factor 1 |
| PBS | phosphate-buffered saline |
| PD | periodontal disease |
| Pg | P. gingivalis W83 stain infection |
| PF | pair-fed |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling assay |
References
- Ma, J.; Guillot, A.; Yang, Z.; Mackowiak, B.; Hwang, S.; Park, O.; Peiffer, B.J.; Ahmadi, A.R.; Melo, L.; Kusumanchi, P.; et al. Distinct histopathological phenotypes of severe alcoholic hepatitis suggest different mechanisms driving liver injury and failure. J. Clin. Investig. 2022, 132, e157780. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Haber, P.; McCaughan, G.W. Alcoholic Liver Disease in Asia, Europe, and North America. Gastroenterology 2016, 150, 1786–1797. [Google Scholar] [CrossRef]
- Asrani, S.K.; Mellinger, J.; Arab, J.P.; Shah, V.H. Reducing the Global Burden of Alcohol-Associated Liver Disease: A Blueprint for Action. Hepatology 2021, 73, 2039–2050. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Younossi, Y.; Golabi, P.; Mishra, A.; Rafiq, N.; Henry, L. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 2020, 69, 564–568. [Google Scholar] [CrossRef]
- Novacek, G.; Plachetzky, U.; Potzi, R.; Lentner, S.; Slavicek, R.; Gangl, A.; Ferenci, P. Dental and periodontal disease in patients with cirrhosis—Role of etiology of liver disease. J. Hepatol. 1995, 22, 576–582. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Lunar Silva, I.; Cascales, E. Molecular Strategies Underlying Porphyromonas gingivalis Virulence. J. Mol. Biol. 2021, 433, 166836. [Google Scholar] [CrossRef]
- Aleksijevic, L.H.; Aleksijevic, M.; Skrlec, I.; Sram, M.; Sram, M.; Talapko, J. Porphyromonas gingivalis Virulence Factors and Clinical Significance in Periodontal Disease and Coronary Artery Diseases. Pathogens 2022, 11, 1173. [Google Scholar] [CrossRef]
- Hussain, M.; Stover, C.M.; Dupont, A.P. gingivalis in Periodontal Disease and Atherosclerosis—Scenes of Action for Antimicrobial Peptides and Complement. Front. Immunol. 2015, 6, 45. [Google Scholar] [CrossRef]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef]
- Ruan, Q.; Guan, P.; Qi, W.; Li, J.; Xi, M.; Xiao, L.; Zhong, S.; Ma, D.; Ni, J. Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front. Immunol. 2023, 14, 1103592. [Google Scholar] [CrossRef]
- Zhou, N.; Zou, F.; Cheng, X.; Huang, Y.; Zou, H.; Niu, Q.; Qiu, Y.; Shan, F.; Luo, A.; Teng, W.; et al. Porphyromonas gingivalis induces periodontitis, causes immune imbalance, and promotes rheumatoid arthritis. J. Leukoc. Biol. 2021, 110, 461–473. [Google Scholar] [CrossRef]
- Ahmadi, P.; Mahmoudi, M.; Kheder, R.K.; Faraj, T.A.; Mollazadeh, S.; Abdulabbas, H.S.; Esmaeili, S.A. Impacts of Porphyromonas gingivalis periodontitis on rheumatoid arthritis autoimmunity. Int. Immunopharmacol. 2023, 118, 109936. [Google Scholar] [CrossRef]
- Zhou, Y.; Vatsalya, V.; Gobejishvili, L.; Lamont, R.J.; McClain, C.J.; Feng, W. Porphyromonas gingivalis as a Possible Risk Factor in the Development/Severity of Acute Alcoholic Hepatitis. Hepatol. Commun. 2019, 3, 293–304. [Google Scholar] [CrossRef]
- Naito, M.; Hirakawa, H.; Yamashita, A.; Ohara, N.; Shoji, M.; Yukitake, H.; Nakayama, K.; Toh, H.; Yoshimura, F.; Kuhara, S.; et al. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 2008, 15, 215–225. [Google Scholar] [CrossRef]
- Igboin, C.O.; Griffen, A.L.; Leys, E.J. Porphyromonas gingivalis strain diversity. J. Clin. Microbiol. 2009, 47, 3073–3081. [Google Scholar] [CrossRef]
- Jiang, M.; Li, F.; Liu, Y.; Gu, Z.; Zhang, L.; Lee, J.; He, L.; Vatsalya, V.; Zhang, H.G.; Deng, Z.; et al. Probiotic-derived nanoparticles inhibit ALD through intestinal miR194 suppression and subsequent FXR activation. Hepatology 2023, 77, 1164–1180. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef]
- Clifford, R.J.; Milillo, M.; Prestwood, J.; Quintero, R.; Zurawski, D.V.; Kwak, Y.I.; Waterman, P.E.; Lesho, E.P.; Mc Gann, P. Detection of bacterial 16S rRNA and identification of four clinically important bacteria by real-time PCR. PLoS ONE 2012, 7, e48558. [Google Scholar] [CrossRef]
- Gravitte, A.; Kintner, J.; Brown, S.; Cobble, A.; Kennard, B.; Hall, J.V. The hormonal environment and estrogen receptor signaling alters Chlamydia muridarum infection in vivo. Front. Cell. Infect. Microbiol. 2022, 12, 939944. [Google Scholar] [CrossRef]
- Cardiff, R.D.; Miller, C.H.; Munn, R.J. Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb. Protoc. 2014, 2014, 655–658. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Albuquerque-Souza, E.; Sahingur, S.E. Periodontitis, chronic liver diseases, and the emerging oral-gut-liver axis. Periodontol. 2000 2022, 89, 125–141. [Google Scholar] [CrossRef]
- Gao, B.; Xu, M.J.; Bertola, A.; Wang, H.; Zhou, Z.; Liangpunsakul, S. Animal Models of Alcoholic Liver Disease: Pathogenesis and Clinical Relevance. Gene Expr. 2017, 17, 173–186. [Google Scholar] [CrossRef]
- Gustot, T.; Fernandez, J.; Szabo, G.; Albillos, A.; Louvet, A.; Jalan, R.; Moreau, R.; Moreno, C. Sepsis in alcohol-related liver disease. J. Hepatol. 2017, 67, 1031–1050. [Google Scholar] [CrossRef]
- Yamanaka, T.; Yamane, K.; Furukawa, T.; Matsumoto-Mashimo, C.; Sugimori, C.; Nambu, T.; Obata, N.; Walker, C.B.; Leung, K.P.; Fukushima, H. Comparison of the virulence of exopolysaccharide-producing Prevotella intermedia to exopolysaccharide non-producing periodontopathic organisms. BMC Infect. Dis. 2011, 11, 228. [Google Scholar] [CrossRef]
- Naruishi, K.; Omori, K.; Maeda, H.; Sonoi, N.; Funakoshi, K.; Hirai, K.; Ishii, M.; Kubo, K.; Kobayashi, H.; Tomiyama, T.; et al. Immune responses to Porphyromonas gingivalis infection suppress systemic inflammatory response in experimental murine model. J. Biol. Regul. Homeost. Agents 2011, 25, 195–202. [Google Scholar]
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007, 46, 831–840. [Google Scholar] [CrossRef]
- Wieser, V.; Tymoszuk, P.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Pfister, A.; Lichtmanegger, L.; Gerner, R.; Drach, M.; et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J. Hepatol. 2016, 64, 872–880. [Google Scholar] [CrossRef]
- Tarantino, G.; Scalera, A.; Finelli, C. Liver-spleen axis: Intersection between immunity, infections and metabolism. World J. Gastroenterol. 2013, 19, 3534–3542. [Google Scholar] [CrossRef]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Roohani, S.; Tacke, F. Liver Injury and the Macrophage Issue: Molecular and Mechanistic Facts and Their Clinical Relevance. Int. J. Mol. Sci. 2021, 22, 7249. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Burns, E.; Eliyahu, T.; Uematsu, S.; Akira, S.; Nussbaum, G. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J. Immunol. 2010, 184, 1455–1462. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Jaeschke, H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol 2002, 27, 23–27. [Google Scholar] [CrossRef]
- Guardado, S.; Ojeda-Juarez, D.; Kaul, M.; Nordgren, T.M. Comprehensive review of lipocalin 2-mediated effects in lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L726–L733. [Google Scholar] [CrossRef]
- Sadik, C.D.; Luster, A.D. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 207–215. [Google Scholar] [CrossRef]
- Xu, M.J.; Feng, D.; Wu, H.; Wang, H.; Chan, Y.; Kolls, J.; Borregaard, N.; Porse, B.; Berger, T.; Mak, T.W.; et al. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: A critical role for IL-6/STAT3. Hepatology 2015, 61, 692–702. [Google Scholar] [CrossRef]
- Olsen, I.; Hajishengallis, G. Major neutrophil functions subverted by Porphyromonas gingivalis. J. Oral Microbiol. 2016, 8, 30936. [Google Scholar] [CrossRef]
- Miralda, I.; Uriarte, S.M.; McLeish, K.R. Multiple Phenotypic Changes Define Neutrophil Priming. Front. Cell. Infect. Microbiol. 2017, 7, 217. [Google Scholar] [CrossRef]
- Ellis, T.N.; Beaman, B.L. Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology 2004, 112, 2–12. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef]
- Gahmberg, C.G. Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr. Opin. Cell Biol. 1997, 9, 643–650. [Google Scholar] [CrossRef]
- Vandendriessche, S.; Cambier, S.; Proost, P.; Marques, P.E. Complement Receptors and Their Role in Leukocyte Recruitment and Phagocytosis. Front. Cell Dev. Biol. 2021, 9, 624025. [Google Scholar] [CrossRef]
- El Kebir, D.; Filep, J.G. Modulation of Neutrophil Apoptosis and the Resolution of Inflammation through beta2 Integrins. Front. Immunol. 2013, 4, 60. [Google Scholar] [CrossRef]
- DeLeo, F.R. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis 2004, 9, 399–413. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Pias, E.K.; Ekshyyan, O.Y.; Rhoads, C.A.; Fuseler, J.; Harrison, L.; Aw, T.Y. Differential effects of superoxide dismutase isoform expression on hydroperoxide-induced apoptosis in PC-12 cells. J. Biol. Chem. 2003, 278, 13294–13301. [Google Scholar] [CrossRef]
- El Kebir, D.; Damlaj, A.; Filep, J.G. Toll-like receptor 9 signaling delays neutrophil apoptosis by increasing transcription of Mcl-1. PLoS ONE 2014, 9, e87006. [Google Scholar] [CrossRef]
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666. [Google Scholar] [CrossRef]
- Kumar, V. The complement system, toll-like receptors and inflammasomes in host defense: Three musketeers’ one target. Int. Rev. Immunol. 2019, 38, 131–156. [Google Scholar] [CrossRef]
- Yu, S.; Wang, D.; Huang, L.; Zhang, Y.; Luo, R.; Adah, D.; Tang, Y.; Zhao, K.; Lu, B. The complement receptor C5aR2 promotes protein kinase R expression and contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages. J. Biol. Chem. 2019, 294, 8384–8394. [Google Scholar] [CrossRef]
- Wu, G.; Zhu, Q.; Zeng, J.; Gu, X.; Miao, Y.; Xu, W.; Lv, T.; Song, Y. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-kappaB. J. Thorac. Dis. 2019, 11, 4816–4828. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef]
- Castro, S.A.; Collighan, R.; Lambert, P.A.; Dias, I.H.; Chauhan, P.; Bland, C.E.; Milic, I.; Milward, M.R.; Cooper, P.R.; Devitt, A. Porphyromonas gingivalis gingipains cause defective macrophage migration towards apoptotic cells and inhibit phagocytosis of primary apoptotic neutrophils. Cell Death Dis. 2017, 8, e2644. [Google Scholar] [CrossRef]
- Yoneda, M.; Hirofuji, T.; Anan, H.; Matsumoto, A.; Hamachi, T.; Nakayama, K.; Maeda, K. Mixed infection of Porphyromonas gingivalis and Bacteroides forsythus in a murine abscess model: Involvement of gingipains in a synergistic effect. J. Periodontal Res. 2001, 36, 237–243. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Ross, R.A.; Crabb, D.W. Activation of carbohydrate response element-binding protein by ethanol. J. Investig Med. 2013, 61, 270–277. [Google Scholar] [CrossRef]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer (FW) | Reverse Primer (RE) |
|---|---|---|
| Actb | 5′-AGACTTCGAGCAGGAGATGG | 5′-CGCTCGTTGCCAATAGTGAT |
| Ccr2 | 5′-GCCTGATCCTGCCTCTACTT | 5′-GGCAAAGATGAGCCTCACAG |
| Ccr5 | 5′-CACACCCTGTTTCGCTGTAG | 5’-ATTCCTGGAAGGTGGTCAGG |
| Ccl2 | 5′-GGCCTGCTGTTCACAGTTGC | 5′-CCTGCTGCTGGTGATCCTCT |
| Myd88 | 5′-CAAGTTTGCACTCAGCCTGT | 5′-AACCGCAGGATACTGGGAAA |
| Il6 | 5′-TCCAGTTGCCTTCTTGGGACT | 5′-GCCTCCGACTTGTGAAGTGGT |
| Tnf | 5′-CCAGCCGATGGGTTGTACCT | 5′-TGACGGCAGAGAGGAGGTTG |
| Il1b | 5′-GGCCTTGGGCCTCAAAGGAA | 5′-GCTTGGGATCCACACTCTCCA |
| Ifng | 5′-CAGGCCATCAGCAACAACAT | 5′-GACCTGTGGGTTGTTGACCT |
| Ncf1 | 5′-CTTCAGACCTATCGGGCCAT | 5′-CGCTTTGTCTTCATCTGGCA |
| Lcn2 | 5′-ATGTCACCTCCATCCTGGTC | 5′-GTGGCCACTTGCACATTGTA |
| C5ar1 | 5′-TCCTGCTGCTGGCTACCATT | 5′-GCTAAGACCCAGGCCACTCC |
| Cybb | 5′-TTGCTGTGCACCATGATGAG | 5′-GGGTGTTCACTTGCAATGGT |
| Sod2 | 5′-CCGAGGAGAAGTACCACGAG | 5′-TAGGGCTCAGGTTTGTCCAG |
| Cd18 | 5′-GCCCTCAACGAGATCACCGA | 5′-CTGGCAGGCCTTCTCCTTGT |
| Pg 16S rRNA | 5′-CTGACACTGAAGCACGAAGG | 5′-CTTAACGCTTTCGCTGTGGA |
| Universal 16S | 5′-ACTCCTACGGGAGGCAGCAGT | 5′-ATTACCGCGGCTGCTGGC |
| 18S rRNA | 5′-CCGGACACGGACAGGATTGA | 5′-GCATGCCAGAGTCTCGTTCG |
| C5ar2 | 5′-CCTGGCTCACAGTGCTCTCA | 5′-TGGTCACCGCACTTTCCTCA |
| Tlr9 | 5′-AGCCTGAGCCACACCAACAT | 5′-GTCACCTTCACCGCTCCTGT |
| Il18 | 5′-TTTCTGGACTCCTGCCTGCT | 5′-TGGAAGGTTTGAGGCGGCTT |
| Hmgb1 | 5′-AATCAAAGGCGAGCATCCTG | 5′-TCAGCTTGGCAGCTTTCTTC |
| Tgfb1 | 5′-CCCGTGGCTTCTAGTGCTGA | 5′-ACAGGATCTGGCCACGGATG |
| ChREBP | 5′-GACAGCGGAGTACATCCTGA | 5′-AAGTTGATGGCAGCGTTGAG |
| Srebf1 | 5′-GCAAGGCCATCGACTACATC | 5′-CTGACACCAGGTCCTTCAGT |
| Fasn | 5′-AAGTTGCCCGAGTCAGAGAA | 5′-TTCCAGACCGCTTGGGTAAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; McClain, C.; Feng, W. Porphyromonas gingivalis Strain W83 Infection Induces Liver Injury in Experimental Alcohol-Associated Liver Disease (ALD) in Mice. Appl. Microbiol. 2024, 4, 620-634. https://doi.org/10.3390/applmicrobiol4020043
Zhou Y, McClain C, Feng W. Porphyromonas gingivalis Strain W83 Infection Induces Liver Injury in Experimental Alcohol-Associated Liver Disease (ALD) in Mice. Applied Microbiology. 2024; 4(2):620-634. https://doi.org/10.3390/applmicrobiol4020043
Chicago/Turabian StyleZhou, Yun, Craig McClain, and Wenke Feng. 2024. "Porphyromonas gingivalis Strain W83 Infection Induces Liver Injury in Experimental Alcohol-Associated Liver Disease (ALD) in Mice" Applied Microbiology 4, no. 2: 620-634. https://doi.org/10.3390/applmicrobiol4020043