
Globally Disseminated Multidrug Resistance Plasmids Revealed by Complete Assembly of Multidrug Resistant Escherichia coli and Klebsiella pneumoniae Genomes from Diarrheal Disease in Botswana
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and Antibiotic Susceptibility Testing
2.2. Whole Genome Sequencing
2.3. Genome Assemblies
2.4. Sequence Analysis, Visualization, and Annotation
3. Results
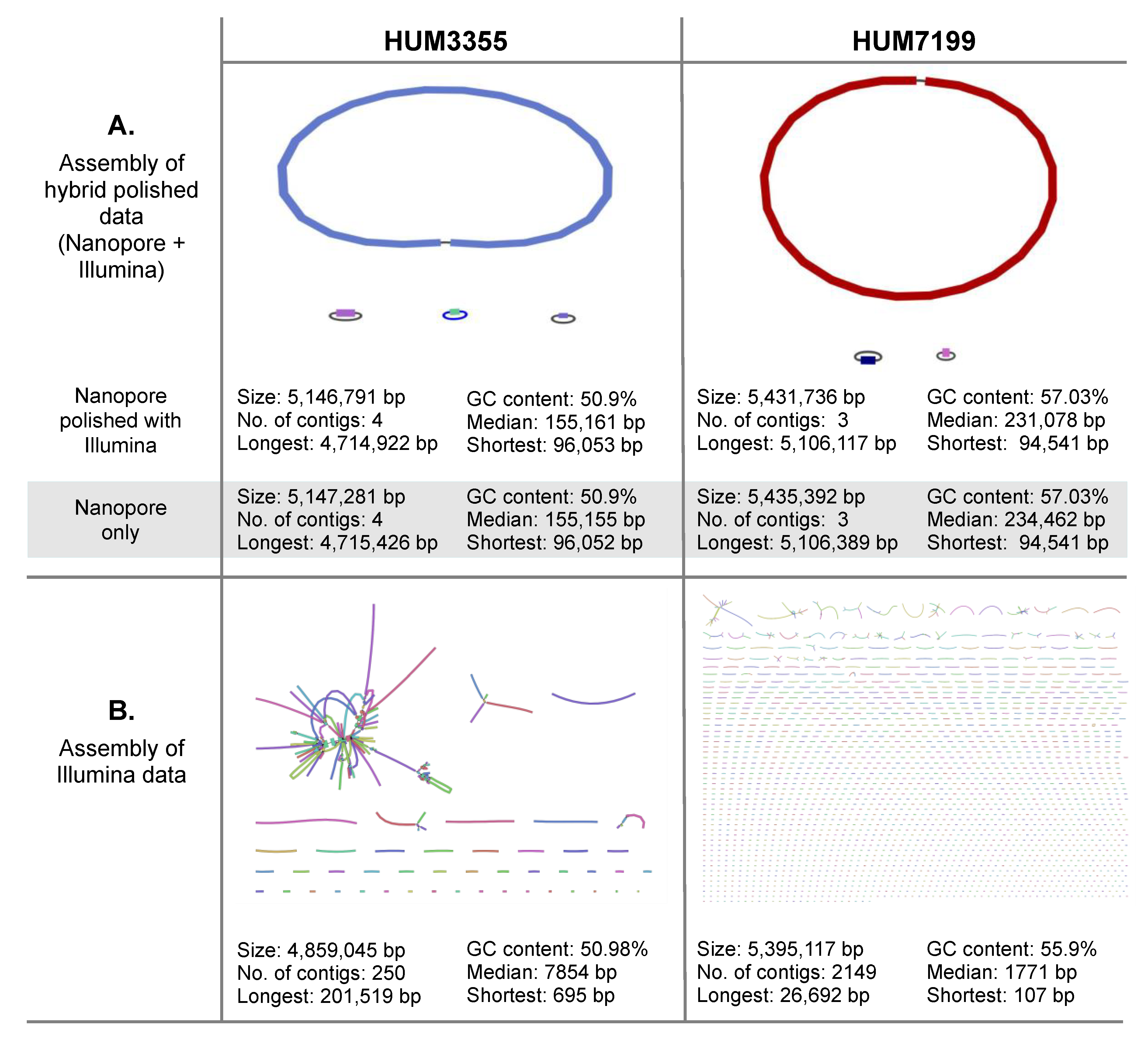
3.1. Comparison of Illumina and Nanopore DNA Sequence Data for Whole Genome Assemblies
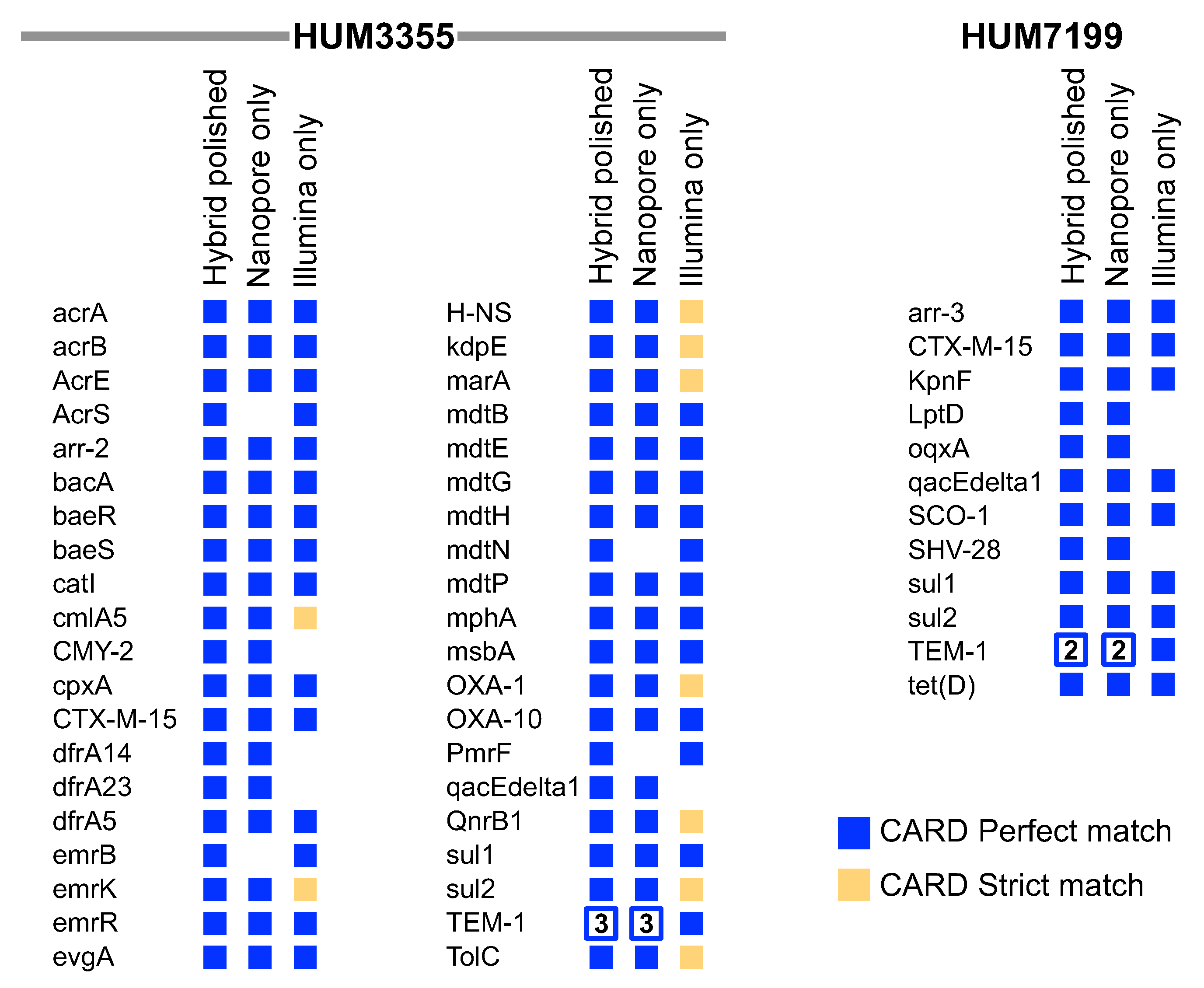
3.2. Whole Genome Comparative Analysis and Detection of Antibiotic Resistance Genes
3.3. Correspondence between Antibiotic Resistance Phenotypes and Predicted ARGs
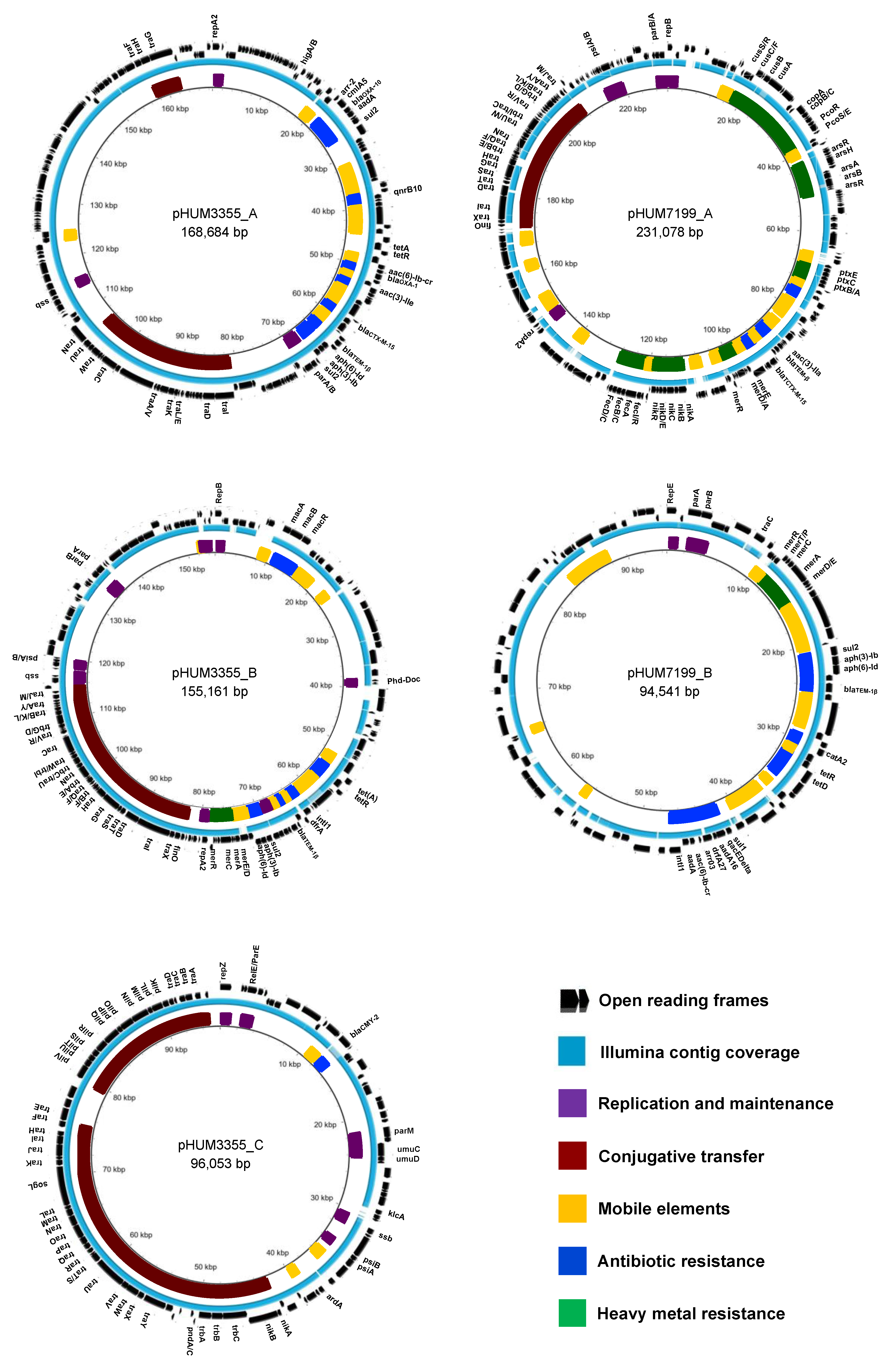
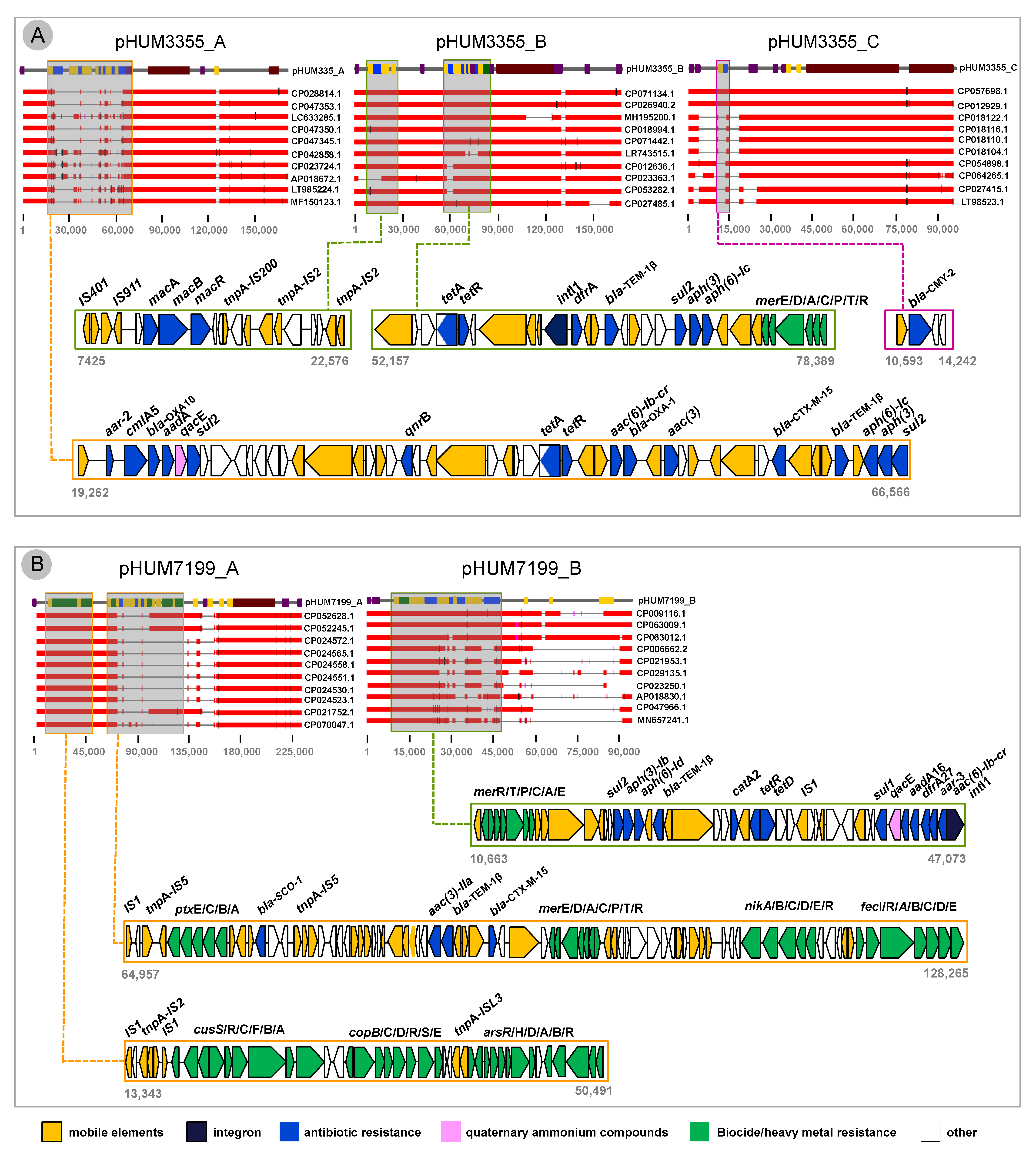
3.4. Plasmid Architecture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; The Review on Antimicrobial Resistance: London, UK, 2014; pp. 1–20. [Google Scholar]
- WHO. Africa Antimicrobial Resistance. Available online: https://www.afro.who.int/health-topics/antimicrobial-resistance (accessed on 10 December 2021).
- Lerminiaux, N.A.; Cameron, A. Horizontal Transfer of Antibiotic Resistance Genes in Clinical Environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mathers, A.J.; Peirano, G.; Pitout, J.D.D. The Role of Epidemic Resistance Plasmids and International High-Risk Clones in the Spread of Multidrug-Resistant Enterobacteriaceae. Clin. Microbiol. Rev. 2015, 28, 565–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antipov, D.; Raiko, M.; Lapidus, A.; Pevzner, P.A. Plasmid Detection and Assembly in Genomic and Metagenomic Data Sets. Genome Res. 2019, 29, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellow, D.; Zorea, A.; Probst, M.; Furman, O.; Segal, A.; Mizrahi, I.; Shamir, R. SCAPP: An Algorithm for Improved Plasmid Assembly in Metagenomes. Microbiome 2021, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Taxt, A.M.; Avershina, E.; Frye, S.A.; Naseer, U.; Ahmad, R. Rapid Identification of Pathogens, Antibiotic Resistance Genes and Plasmids in Blood Cultures by Nanopore Sequencing. Sci. Rep. 2020, 10, 7622. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Masim, M.A.L.; Gayeta, J.M.; Lagrada, M.L.; Macaranas, P.K.V.; Cohen, V.; Limas, M.T.; Espiritu, H.O.; Palarca, J.C.; Chilam, J.; et al. Integrating Whole-Genome Sequencing within the National Antimicrobial Resistance Surveillance Program in the Philippines. Nat. Commun. 2020, 11, 2719. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- NIHR. Global Health Research Unit on Genomic Surveillance of AMR Whole-Genome Sequencing as Part of National and International Surveillance Programmes for Antimicrobial Resistance: A Roadmap. BMJ Glob. Health 2020, 5, e002244. [Google Scholar] [CrossRef]
- Tuan, V.P.; Narith, D.; Tshibangu-Kabamba, E.; Dung, H.D.Q.; Viet, P.T.; Sokomoth, S.; Binh, T.T.; Sokhem, S.; Tri, T.D.; Ngov, S.; et al. A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates. J. Clin. Med. 2019, 8, 858. [Google Scholar] [CrossRef] [Green Version]
- Hanass-Hancock, J.; Carpenter, B. Trends in Health and Disability in Botswana. An Analysis of the Global Burden of Disease Study. Disabil. Rehabil. 2021, 43, 3606–3612. [Google Scholar] [CrossRef]
- Jobbins, S.E.; Alexander, K.A. From Whence They Came—Antibiotic-Resistant Escherichia coli in African Wildlife. J. Wildl. Dis. 2015, 51, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.E.; Fox, J.T.; Dougherty, E.R.; Cameron, A.; Alexander, K.A. The Changing Face of Water: A Dynamic Reflection of Antibiotic Resistance across Landscapes. Front. Microbiol. 2018, 9, 1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapela, K.; Rahube, T. Isolation and Antibiotic Resistance Profiles of Bacteria from Influent, Effluent and Downstream: A Study in Botswana. Afr. J. Microbiol. Res. 2019, 13, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Lesho, E.; Clifford, R.; Onmus-Leone, F.; Appalla, L.; Snesrud, E.; Kwak, Y.; Ong, A.; Maybank, R.; Waterman, P.; Rohrbeck, P.; et al. The Challenges of Implementing Next Generation Sequencing Across a Large Healthcare System, and the Molecular Epidemiology and Antibiotic Susceptibilities of Carbapenemase-Producing Bacteria in the Healthcare System of the U.S. Department of Defense. PLoS ONE 2016, 11, e0155770. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.; Blackburn, J. Overcoming Barriers in Evaluating Outbreaks of Diarrheal Disease in Resource Poor Settings: Assessment of Recurrent Outbreaks in Chobe District, Botswana. BMC Public Health 2013, 13, 775. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.A.; Herbein, J.; Zajac, A. The Occurrence of Cryptosporidium and Giardia Infections Among Patients Reporting Diarrheal Disease in Chobe District, Botswana. Adv. Infect. Dis. 2012, 2, 137–147. [Google Scholar] [CrossRef]
- Alexander, K.A.; Heaney, A.K.; Shaman, J. Hydrometeorology and Flood Pulse Dynamics Drive Diarrheal Disease Outbreaks and Increase Vulnerability to Climate Change in Surface-Water-Dependent Populations: A Retrospective Analysis. PLOS Med. 2018, 15, e1002688. [Google Scholar] [CrossRef] [Green Version]
- Pesapane, R.; Ponder, M.; Alexander, K.A. Tracking Pathogen Transmission at the Human–Wildlife Interface: Banded Mongoose and Escherichia coli. EcoHealth 2013, 10, 115–128. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus Long-Read Assemblies for Bacterial Genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Šikić, M. Time- and Memory-Efficient Genome Assembly with Raven. Nat. Comput. Sci. 2021, 1, 332–336. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap and Miniasm: Fast Mapping and de Novo Assembly for Noisy Long Sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; O’Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.K.; Durkin, A.S.; et al. RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline Reach with Protein Family Model Curation. Nucleic Acids Res. 2021, 49, D1020–D1028. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST. Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aasrestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Garcillán-Barcia, M.P.; Redondo-Salvo, S.; Vielva, L.; de la Cruz, F. MOBscan: Automated Annotation of MOB Relaxases. Methods Mol. Biol. 2020, 2075, 295–308. [Google Scholar] [CrossRef]
- Redondo-Salvo, S.; Bartomeus-Peñalver, R.; Vielva, L.; Tagg, K.A.; Webb, H.E.; Fernández-López, R.; de la Cruz, F. COPLA, a Taxonomic Classifier of Plasmids. BMC Bioinform. 2021, 22, 390. [Google Scholar] [CrossRef]
- Kary, S.C.; Yoneda, J.; Olshefsky, S.C.; Stewart, L.A.; West, S.B.; Cameron, A. The Global Regulatory Cyclic AMP Receptor Protein (CRP) Controls Multifactorial Fluoroquinolone Susceptibility in Salmonella enterica Serovar Typhimurium. Antimicrob. Agents Chemother. 2017, 61, e01666-17. [Google Scholar] [CrossRef] [Green Version]
- Law, A.; Solano, O.; Brown, C.J.; Hunter, S.S.; Fagnan, M.; Top, E.M.; Stalder, T. Biosolids as a Source of Antibiotic Resistance Plasmids for Commensal and Pathogenic Bacteria. Front. Microbiol. 2021, 12, 606409. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Defining and Combating Antibiotic Resistance from One Health and Global Health Perspectives. Nat. Microbiol. 2019, 4, 1432–1442. [Google Scholar] [CrossRef]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Antibiotic Resistance: Moving from Individual Health Norms to Social Norms in One Health and Global Health. Front. Microbiol. 2020, 11, 1914. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, P.M.C.; Flach, C.-F.; Larsson, D.G.J. A Conceptual Framework for the Environmental Surveillance of Antibiotics and Antibiotic Resistance. Environ. Int. 2019, 130, 104880. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R. A One-Health Approach to Antimicrobial Resistance. Nat. Microbiol. 2018, 3, 854–855. [Google Scholar] [CrossRef]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. EcoHealth 2019, 16, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.-G. Using the Class 1 Integron-Integrase Gene as a Proxy for Anthropogenic Pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Huguet, K.T.; Coulombe, F.; Burrus, V. Entry Exclusion of Conjugative Plasmids of the IncA, IncC, and Related Untyped Incompatibility Groups. J. Bacteriol. 2019, 201, e00731-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macori, G.; Nguyen, S.V.; Naithani, A.; Hurley, D.; Bai, L.; El Garch, F.; Woehrlé, F.; Miossec, C.; Roques, B.; O’Gaora, P.; et al. Characterisation of Early Positive Mcr-1 Resistance Gene and Plasmidome in Escherichia coli Pathogenic Strains Associated with Variable Phylogroups under Colistin Selection. Antibiotics 2021, 10, 1041. [Google Scholar] [CrossRef]
- Kudirkiene, E.; Andoh, L.A.; Ahmed, S.; Herrero-Fresno, A.; Dalsgaard, A.; Obiri-Danso, K.; Olsen, J.E. The Use of a Combined Bioinformatics Approach to Locate Antibiotic Resistance Genes on Plasmids from Whole Genome Sequences of Salmonella enterica Serovars from Humans in Ghana. Front. Microbiol. 2018, 9, 1010. [Google Scholar] [CrossRef]
- Ajayi, A.O.; Perry, B.J.; Yost, C.K.; Jamieson, R.C.; Hansen, L.T.; Rahube, T.O. Comparative Genomic Analyses of β-Lactamase (bla CMY-42)-Encoding Plasmids Isolated from Wastewater Treatment Plants in Canada. Can. J. Microbiol. 2021, 67, 737–748. [Google Scholar] [CrossRef]
- Tian, D.; Wang, M.; Zhou, Y.; Hu, D.; Ou, H.-Y.; Jiang, X. Genetic Diversity and Evolution of the Virulence Plasmids Encoding Aerobactin and Salmochelin in Klebsiella pneumoniae. Virulence 2021, 12, 1323–1333. [Google Scholar] [CrossRef]
- Jing, Y.; Jiang, X.; Yin, Z.; Hu, L.; Zhang, Y.; Yang, W.; Yang, H.; Gao, B.; Zhao, Y.; Zhou, D.; et al. Genomic Diversification of IncR Plasmids from China. J. Glob. Antimicrob. Resist. 2019, 19, 358–364. [Google Scholar] [CrossRef] [PubMed]
- García, P.; Hopkins, K.L.; García, V.; Beutlich, J.; Mendoza, M.C.; Threlfall, J.; Mevius, D.; Helmuth, R.; Rodicio, M.R.; Guerra, B.; et al. Diversity of Plasmids Encoding Virulence and Resistance Functions in Salmonella enterica Subsp. Enterica Serovar Typhimurium Monophasic Variant 4,[5],12:I:− Strains Circulating in Europe. PLoS ONE 2014, 9, e89635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozwandowicz, M.; Brouwer, M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids Carrying Antimicrobial Resistance Genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Class | Antibiotic Disc * (Phenotype) | Gene Predicted to Confer Resistance ** | ||||
|---|---|---|---|---|---|---|
| Chromosome (4,714,316 bp) | pHUM3355_A (168,684 bp) | pHUM3355_B (155,161 bp) | pHUM3355_C (96,053 bp) | |||
| Aminoglycoside | G10 N30 S10 | (R) (R) (R) | aph(3″)-Ib, aph(6)-Id, mdf(A) | aadA1, aph(3″)-Ib, aph(6)-Id, aac(3)-IIa, aac(6′)-Ib-cr | aph(6)-Id | — |
| Beta-lactam | AMP10 XNL30 | (R) (R) | blaTEM-1β | blaTEM-1β blaCTX-M-15, blaOXA1, blaOXA10 | blaTEM-1β | blaCMY-2 |
| Folate pathway antagonist | SXT25 | (R) | drfA14, sul2 | drfA23, sul1, sul2 | drfA5, sul2 | — |
| Fluoroquinolone | CIP5 | (S) | mdf(A) | qnrB1, aac(6′)-Ib-cr | — | — |
| Macrolide | not tested | mph(A), mdf(A) | mac(A), mac(B) | — | — | |
| Phenicol | C30 | (R) | catA1, mdf(A) | catB3, cmlA1 | — | — |
| Rifamycin | not tested | — | arr2 | — | — | |
| Tetracycline | D30 T30 | (R) (R) | tet(B), mdf(A) | tet(A) | tet(A) | — |
| Quaternary ammonium compounds | not tested | — | qacEDelta | — | — | |
| Antimicrobial Class | Antibiotic Disc * (Phenotype) | Gene Predicted to Confer Resistance ** | |||
|---|---|---|---|---|---|
| Chromosome (5,105,496 bp) | pHUM7199_A (231,078 bp) | pHUM7199_B (94,541 bp) | |||
| Aminoglycoside | G10 N30 S10 | (R) (S) (R) | — | aac(3)-IIa | aadA16, aph(3”)-Ib, aph(6)-Id, aac(6′)-Ib-cr |
| Beta-lactam | AMP10 XNL30 | (R) (R) | blaSHV-106/28 | blaTEM-1β blaSCO-1, blaCTX-M-15 | blaTEM-1β |
| Folate pathway antagonist | SXT25 | (R) | — | — | drfA27, sul1, sul2 |
| Fluoroquinolone | CIP5 | (R) | oqxA, oqxB | — | — |
| Macrolide | not tested | — | — | — | |
| Phenicol | C30 | (R) | — | — | catA2 |
| Rifamycin | not tested | — | — | aar3 | |
| Tetracycline | D30 T30 | (R) (R) | — | — | tet(D) |
| Quaternary ammonium compounds | not tested | — | — | qacEDelta | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahube, T.O.; Cameron, A.D.S.; Lerminiaux, N.A.; Bhat, S.V.; Alexander, K.A. Globally Disseminated Multidrug Resistance Plasmids Revealed by Complete Assembly of Multidrug Resistant Escherichia coli and Klebsiella pneumoniae Genomes from Diarrheal Disease in Botswana. Appl. Microbiol. 2022, 2, 934-949. https://doi.org/10.3390/applmicrobiol2040071
Rahube TO, Cameron ADS, Lerminiaux NA, Bhat SV, Alexander KA. Globally Disseminated Multidrug Resistance Plasmids Revealed by Complete Assembly of Multidrug Resistant Escherichia coli and Klebsiella pneumoniae Genomes from Diarrheal Disease in Botswana. Applied Microbiology. 2022; 2(4):934-949. https://doi.org/10.3390/applmicrobiol2040071
Chicago/Turabian StyleRahube, Teddie O., Andrew D. S. Cameron, Nicole A. Lerminiaux, Supriya V. Bhat, and Kathleen A. Alexander. 2022. "Globally Disseminated Multidrug Resistance Plasmids Revealed by Complete Assembly of Multidrug Resistant Escherichia coli and Klebsiella pneumoniae Genomes from Diarrheal Disease in Botswana" Applied Microbiology 2, no. 4: 934-949. https://doi.org/10.3390/applmicrobiol2040071