
ROS1-Rearranged Lung Adenocarcinoma: From Molecular Genetics to Target Therapy

Abstract
:Simple Summary
Abstract
1. Introduction
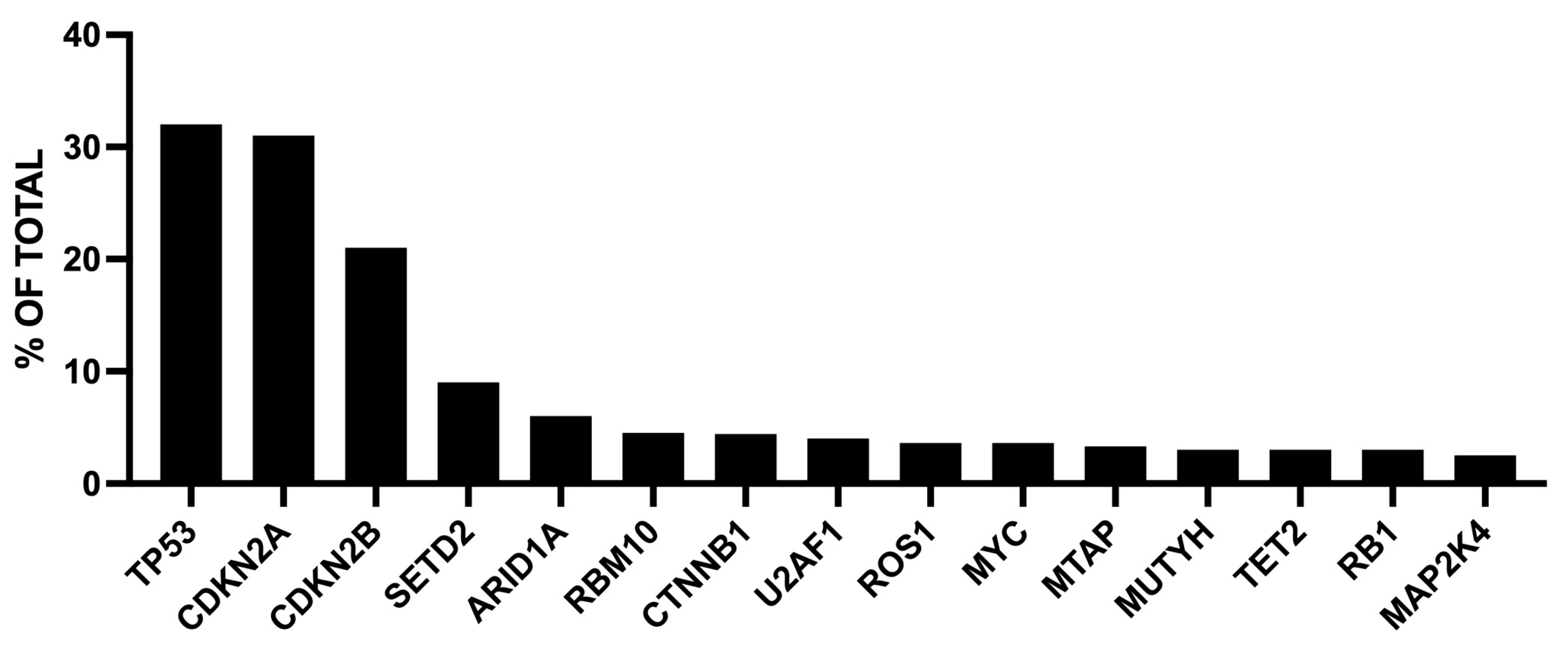
1.1. ROS1 Gene Rearrangements
1.2. Target Therapy of ROS1-Rearranged NSCLC
1.2.1. Crizotinib
1.2.2. Entrectinib
1.2.3. Lorlatinib
1.2.4. Taletrectinib

{kind=link}
| TKI | Clinical Trial Phase | Number of Patients | ORR (%) | DCR (%) | mPFS (mo) | mDOR (mo) | mOS (mo) | Grade ¾ Adverse Events | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Crizotinib | PROFILE 1001 I/II | 53 | 72 | 90 | 19.3 | 24.7 | 51.4 | 36 | [23,24] |
| Crizotinib | East Asian NCT01945021 II | 127 | 72 | 90 | 16 | 19.7 | 44.2 | 25 | [27,28] |
| Crizotinib | Ac Sé I/II | 36 | 47 | 83 | 6 | ------- | 17 | ------- | [25] |
| Crizotinib | EUCROSS II | 34 | 70 | 90 | 20 | ------- | NR | 20 | [26] |
| Crizotinib Platinum (PL) | Retrospective Study | 104 56 (Cr) 46 (Pl) | 84(Cr) 57 (Pl) | 96 (CR) 100 (Pl) | 14.9 (Cr) 8.5 (Pl) | -------- | NR (Cr) NR (Pl) | -------- | [29] |
| Entrectinib | ALKA-372-001 STRATRK-1 STRATRK-2 I/II | 161 | 68 | 90 | 16 | 20.5 | 47.8 | 31 | [33,34,35] |
| Lorlatinib | NCT01970845 I/II | 69 21 (TN) 48 (TT) | 41 (overall) 62 (CN) 35 (CR) | --------- | 25.3 (CN) 13.8 (CR) | --------- | ---------- | 43 | [36] |
| Lorlatinib | LORLATINU | 80 100% (TT) 69% (CT) 64% (BM) | 45 | 82 | 7.1 | 6.9 | 19.6 | 33 | [37] |
| Taletrectinib | TRUST-I | 109 67 (TN) 42 (TT) | 92.5 (TN) 52.6 (TT) | 95.5 (TN) 81.6 (TT) | 33.2 (TN) 9.8 (TT) | NR (TN) NR (TT) | --------- --------- | 29 | [39,40] |
1.2.5. Repotrectinib
1.2.6. NVL-520
1.3. Resistance Mechanisms
1.3.1. Resistance to Crizotinib
1.3.2. Resistance to Lorlatinib
1.3.3. ROS1-Extrinsic Mechanisms
1.3.4. Intracranial Failure
1.3.5. Off-Target Activation of Signaling Pathways
1.3.6. Histological Transformation into Small Cell Lung Cancer (SCLC) of ROS1-Rearranged NSCLC
2. Limitations of the Clinical Studies Carried Out on ROS1-Rearranged LUADs
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harada, G.; Yang, S.R.; Cocco, E.; Drilon, A. Rare molecular subtypes of lung cancer. Nat. Rev. Clin. Oncol. 2023, 20, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G. Molecular characterization of lung adenocarcinoma combining whole genome sequencing, copy number analysis and gene expression profiling. Exp. Rev. Mol. Diagn. 2022, 22, 77–100. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Lavery, J.A.; Brown, S.; de Bruijn, I.; Jee, J.; Tran, T.N.; Rizvi, H.; Arbour, K.C.; Whiting, K.; Shen, R.; et al. The GENIE BPC NSCLCX cohort: A real-world repository integrating standardized clinical and genomic data for 1846 patients with non-small cell lung cancer. Clin. Cancer Res. 2023, in press. [Google Scholar]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9270–9274. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Kaack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehora, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–382. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, S.R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Incarbone, M.; Roncalli, M.; Cappuzzo, F.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, C.; Ding, W.; Zhang, Z.; Qiu, X.; Mu, D.; Zhang, H.; Xi, Y.; Zhou, J.; Ma, L.; et al. Prevalence of ROS1 fusion in Chinese patients with non-small cell lung cancer. Thorac. Cancer 2019, 10, 47–53. [Google Scholar] [CrossRef]
- Kim, H.R.; Lim, S.M.; Kim, H.J.; Hwang, S.K.; Park, J.K.; Shin, E.; Bae, M.K.; Ou, S.H.I.; Wang, J.; Jewell, S.S.; et al. The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann. Oncol. 2013, 24, 2364–2368. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, P.L.; Kim, H.; Park, E.; Shim, H.S.; Jheon, S.; Kim, K.; Lee, C.T.; Chung, J.H. ROS1 gene rearrangement and copy number gain in non-small cell lung cancer. Wichows Arch. 2015, 466, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Ritterhouse, L.L.; Siraj, M. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J. Thoracic. Oncol. 2017, 5, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhao, C.; Li, J.; Su, C.; Chen, X.; Ren, S.; Li, X.; Zhou, C. Clinical features, and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019, 8, 2858–2866. [Google Scholar] [CrossRef]
- Huang, R.; Haberberger, J.; Sokol, E.; Schrock, A.B.; Danziger, N.; Madison, R.; Trabucco, R.; Jin, D.; Pavlick, D.; Ramanan, V.; et al. Clinicopathologic, genomic and protein expression characterization of 356 ROS1 fusion driven solid tumor cases. Int. J. Cancer 2021, 148, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Schoenfeld, A.J.; Siau, E.; Lu, Y.C.; Tai, H.; Suzawa, K.; Kubota, D.; Lui, A.; Qerigi, B.; Mattar, M.; et al. MAPK pathway alterations correlate with poor survival and drive resistance to the therapy in patients with lung cancers driven by ROS1 fusions. Clin. Cancer Res. 2020, 26, 2932–2945. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, H.; Lu, X.; Wang, W.; Chen, H.; Robinson, M.K.; Chang, J.; Tang, G.; Medeiros, L.J. Coexistent genetic alterations involving ALK, RET, ROS1 or MET in 15 cases of lung adenocarcinoma. Mod. Pathol. 2018, 31, 307–312. [Google Scholar] [CrossRef]
- Gendarme, S.; Bylioski, D.; Chouaid, L.; Guiser, F. ROS-1 fusions in non-small-cell lung cancer: Evidence to date. Curr. Oncol. 2022, 29, 641–658. [Google Scholar] [CrossRef]
- Jun, H.J.; Johnson, H.; Bronson, R.T.; de Feraudy, S.; White, F.; Charest, A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012, 72, 3764–3774. [Google Scholar] [CrossRef]
- Roskoski, R. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol. Res. 2017, 121, 202–212. [Google Scholar] [CrossRef]
- Neel, D.S.; Allegakoen, D.V.; Olivas, V.; Mayekar, M.K.; Hemmati, G.; Chatterejee, N.; Blakely, C.M.; McCoach, C.E.; Rootw, J.K.; Le, A.; et al. Differential subcellular localization regulates oncogenic signaling by ROS1 kinase fusion proteins. Cancer Res. 2019, 79, 546–556. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Takahashi, H.; Nakamura, H.; Hama, N.; Kohno, T.; Tsuta, K.; Yoshida, A.; Asamura, H.; Mutoh, M.; et al. Mouse model for ROS1-rearranged lung cancer. PLoS ONE 2013, 8, e56010. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Toki, H.; Matsui, J.; Togashi, Y.; Dobashi, A.; Fukumura, R.; Gondo, Y.; Monowa, O.; Tanaka, N.; Mori, S.; et al. Mouse models for ROS1-fusion-positive lung cancers and their application to the analysis of multikinase inhibitor efficiency. Carcinogenesis 2016, 37, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Uo, S.H.I.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef]
- Shaw, A.T.; Riely, G.; Bang, Y.J.; Kim, D.W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.; Shapiro, G.; Usari, T.; et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Moro-Sibilot, D.; Cozic, N.; Pérol, M.; Mazières, J.; Otto, J.; Souquet, P.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSé phase II trial. Ann. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef]
- Michels, S.; Massutí, B.; Schildhaus, H.-U.; Franklin, J.; Sebastian, M.; Felip, E.; Grohé, C.; Rodriguez-Abreu, D.; Abdulla, D.S.; Bischoff, H.; et al. Safety and Efficacy of Crizotinib in Patients With Advanced or Metastatic ROS1-Rearranged Lung Cancer (EUCROSS): A European Phase II Clinical Trial. J. Thorac. Oncol. 2019, 14, 1266–1276. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Yang, J.C.-H.; Kim, D.-W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.-J.; Yamamoto, N.; Ahn, M.-J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients with ROS1-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef]
- Wu, J.L.; Lu, S.; Yang, J.; Zhou, J.; Seto, T.; Ahn, M.J.; Su, W.C.; Yamamoto, N.; Kim, D.W.; Paolini, J.; et al. Final overall survival, safety, and quality of life. Results from a phase 2 study of crizotinib in East Asian patients with ROS1-positive advanced NSCLC. JTO Clin. Res. Rep. 2022, 3, 100406. [Google Scholar] [CrossRef]
- Shen, L.; Qiang, T.; Li, Z.; Ding, D.; Yu, Y.; Lu, S. First-line crizotinib versus platinum-pemetrexed chemotherapy in patients with advanced ROS1-rearranged non-small-cell lung cancer. Cancer Med. 2020, 9, 3310–3318. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Q.; Liang, L.; Li, J.; Liu, Z.; Li, W.; Yang, L.; Yang, G.; Xu, F.; Ying, J.; et al. Crizotinib vs platinum-based chemotherapy as first-line treatment for advanced noin-small cell lung cancer with different ROS1 fusion variants. Cancer Med. 2020, 9, 3328–3336. [Google Scholar] [CrossRef]
- Krawczyk, P.; Grenda, A.; Terelcka, P.; Blach, J.; Wojas-Krawczyk, K.; Kuckarczyck, T.; Chmielewska, I.; Kiesko, R.; Jarosz, B.; Gil, M.; et al. Crizotinib efficacy in advanced non-squamous NSCLC patients with ALK or ROS1 rearrangement. Sci. Rep. 2021, 11, 20939. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Zhang, R.; Xu, Q.; Yang, H.; Lizaso, A.; Xu, C.; Liu, J.; Wang, W.; Ou, S.H.I.; et al. Clinical and molecular factors that impact the efficacy of first-line crizotinib in ROS1-rearranged non-small-cell lung cancer: A large multicenter retrospective study. BMC Med. 2021, 19, 206. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; De Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Krebs, M.G.; De Braud, F.; Siena, S.; Drilon, A.; Doebele, R.C.; Patel, M.R.; Cho, B.C.; Liu, S.V.; Ahn, M.J.; et al. Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small-cell lung cancer. J. Clin. Oncol. 2021, 39, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Chu, C.H.; Fan, Y.; Cho, B.C.; Lu, S.; Ahn, M.J.; Krebs, M.G.; Liu, S.V.; John, T.; Otterson, G.A.; et al. Long-term efficacy and safety of entretectinib in ROS1 fusion-positive NSCLC. JTO Clin. Res. Rep. 2022, 3, 100332. [Google Scholar]
- Shaw, A.T.; Solomon, B.J.; Chiari, R.; Riely, G.J.; Besse, B.; Soo, R.A.; Kao, S.; Lin, C.C.; Bauer, T.M.; Clancy, J.S.; et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2019, 20, 1691–1701. [Google Scholar] [CrossRef]
- Girard, N.; Galland-girodet, S.; Avrillon, V.; Besse, B.; Duruisseaux, M.; Cadranel, J.; Otto, J.; Prevost, A.; Roch, B.; Bennouna, J.; et al. Lorlatinib for advanced ROS1+ non-small-cell lung cancer: Results of the IFCT-1803 LORLATU study. ESMO Open Cancer Horiz. 2022, 7, 100418. [Google Scholar] [CrossRef]
- Ou, D.S.H.I.; Fuijiwara, Y.; Shaw, A.T.; Yamamoto, N.; Nakagawa, K.; Fan, F.; Hao, Y.; Gao, Y.; Janne, P.A.; Seto, T. Efficacy of taletrectinib (AB-106/DS-6051b) in ROS1+ NSCLC: An updataed pooled analysis of U.S. anf Japan phase 1 studies. JTO Clin. Res. Rep. 2020, 2, 100108. [Google Scholar]
- Li, W.; Yang, N.; Ma, H.; Fan, H.; Li, K.; Wu, H.; Yu, Q.; Wang, Y.; Meng, X.; Wang, X.; et al. The efficacy and safety of taletrectinib in patients with TKI-naïve or crizotinib-pretreated ros1-positive non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2023, 40 (Suppl. S16), 8572. [Google Scholar] [CrossRef]
- Li, W.; Yang, N.; Li, K.; Fan, H.; Yu, Q.; Wu, H.; Wang, Y.; Meng, X.; Wu, J.; Wang, Z.; et al. Updated efficacy and safety of taletrectinib in patients (pts) with ROS1+ non-small cell lung cancer (NSCLC). J. Thoracic. Oncol. 2023, 18, S47–S48. [Google Scholar] [CrossRef]
- Nagasawa, M.; Ohe, Y.; Zhou, C.; Choi, C.; Yang, N.; Liu, G.; Felip, R.; Pérol, M.; Besse, B.; Nieva, J.; et al. TRUST-II a global phase II study of taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors. Future Oncol. 2023, 19, 123–135. [Google Scholar]
- Yun, M.R.; Kim, D.H.; Jo, H.S.; Lee, Y.W.; Choi, H.M.; Park, C.W.; Heo, S.G.; Kang, H.N.; Lee, S.S.; Schoenfeld, A.J.; et al. Repotrectinib exhibits potent antitumor activity in treatment-naïve and solvent-front-mutant ROS1-rearranged non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Drilon, A.E.; Cho, B.C.; Felip, E.; De Langen, A.; Yang, N. Intracranial and systemic efficacy of repotrectinib in advanced ROS1 fusion-positive (ROS1+) non-small cell lung cancer (NSCLC) and central nervous system metastases (CNS mets) in the phase 1-2 TRIDENT-1. J. Clin. Oncol. 2023, 41 (Suppl. S16), 9017. [Google Scholar] [CrossRef]
- Cho, B.C.; Lin, J.; Camidge, D.R.; Velcheti, V.; Solomon, B.; Lu, S.; Lee, K.H.; Kim, S.W.; Kao, S.; Diadziuskzko, R.; et al. Pivotal topline data from the phase 1-2 TRIDENT-1 trial of repotrectinib in patients with ROS1+ advanced non-small cell lung cancer (NSCLC). Eur. J. Cancer 2022, 174, S1–S2. [Google Scholar] [CrossRef]
- Drilon, A.; Horan, J.C.; Tangpeerachaikul, A.; Besse, B.; Ou, S.H.I.; Gadgeel, S.M.; Camidge, D.R.; van der Wekken, A.; Nguyen-Phuoing, L.; Acker, A.; et al. NVL-520 is a selective, TRK-sparing, and brain-penetrant inhibitor of ROS1 fusions and secondary resistance mutations. Cancer Discov. 2023, 13, 598–615. [Google Scholar] [CrossRef]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef]
- Gou, W.; Zhou, X.; Liu, Z.; Wang, L.; Shen, J.; Xu, X.; Li, Z.; Zhai, X.; Zuo, D.; Wu, Y. CD74-ROS1 G2032R mutation transcriptionally up-regulates Twist1 in non-small cell lung cancer cells leading to increased migration, invasion, and resistance to crizotinib. Cancer Lett. 2018, 422, 19–28. [Google Scholar] [CrossRef]
- Song, A.; Kim, T.M.; Kim, D.W.; Keam, B.; Lee, S.H.; Heo, D.S. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin. Cnacer Res. 2015, 21, 2379–2387. [Google Scholar] [CrossRef]
- Facchinetti, F.; Loriot, Y.; Cossin-Kuo, M.S.; Mahjoubi, L.; Lacroix, L.; Planchard, D.; Besse, B.; Farace, F.; Auger, N.; Remon, J.; et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin. Cancer Res. 2016, 22, 5983–5991. [Google Scholar] [CrossRef]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol. 2017, 2017, PO.17.00063. [Google Scholar] [CrossRef]
- Lin, J.J.; Choudhury, N.J.; Yoda, S.; Zhu, V.W.; Johnson, T.W.; Sakhtemani, R.; Dagogo-Jack, I.; Digumathy, S.R.; Lee, C.; Do, A.; et al. Spectrum of mechanisms of resistance to crizotinib and lorlatinib in ROS1 fusion-positive lung cancer. Clin. Cancer Res. 2021, 27, 2899–2909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Z.; Zeng, L.; Zhang, X.; Li, Y.; Xu, Q.; Yang, H.; Lizaso, A.; Xu, C.; Liu, J.; et al. Disease progression patterns and molecular resistance mechanisms to crizotinib of lung adenocarcinoma harboring ROS1 rearrangements. NPJ. Precis. Oncol. 2022, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, X.; Chen, H.; Bao, H.; Wu, X.; Wang, H.; Bao, H.; Pang, J.; Wang, S.; Tang, H.; et al. Mechanisms of resistance to tyrosine kinase inhibitor treatmenrts in patients with ROS1 fusion-positive non-small cell lung cancer. J. Clin. Oncol. 2022, 41 (Suppl. S16), 9062. [Google Scholar] [CrossRef]
- Patil, T.; Smith, D.E.; Bunn, P.A.; Aisner, D.L.; Le, A.; Hancock, M.; Purcell, W.T.; Bowles, D.W.; Camidge, D.R.; Doebele, R.C. The incidence of brain metastases in stage IV ROS1-rearranged non-small cell lung cancer and rate of central nervous system progression on crizotinib. J. Thorac. Oncol. 2018, 13, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.I.; Zhu, V. CNS metastasis in ROS1+ NSCLC: An urgent call to action, to understand, and to overcome. Lung Cancer 2019, 130, 201–207. [Google Scholar] [CrossRef]
- Okimoto, T.; Tsubata, Y.; Hotta, T.; Hamaguchi, M.; Nakao, M.; Hamaguchi, S.I.; Hamada, A.; Isobe, T. A low crizotinib concentration in the cerebrospinal fluid causes ineffective treatment of anaplastic lymphoma kinase-positive non-small cell lung cancer with carcinomatous meningitis. Intern. Med. 2019, 58, 703–705. [Google Scholar] [CrossRef]
- Gen, L.; Xu, H.; Zhao, J.; Kong, J.; Ai, X.; Yu, F.; Du, K.; Zhu, L.; Ma, H.; Wang, Q.; et al. Concurrent TP53 mutation adversely impact the efficacy of crizotimib in ROS1-rearranged lung cancer patients. J. Clin. Oncol. 2019, 37 (Suppl. S15), e20535. [Google Scholar] [CrossRef]
- Frost, N.; Christopoulos, P.; Kauffmann-Guerrero, D.; Stratmann, J.; Riedel, R.M.; Schefer, M.; Alt, J.; Gutz, S.; Christoph, D.C.; Laack, E.; et al. Lorlatinib in pretreated ALK- or ROS1-positive lung cancer and impact of TP53 co-mutations: Results from the German early access program. Ther. Adv. Med. Oncol. 2021, 13, 1758835920980558. [Google Scholar] [CrossRef]
- Mc Coach, C.E.; Le, A.T.; Gowan, K.; Jones, K.; Schubert, L.; Doak, A.; Estrada-Bernal, A.; Davies, K.D.; Merrick, D.T.; Bunn, P.A.; et al. Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non-small cell lung cancer. Clin. Cancer Res. 2018, 24, 3334–3347. [Google Scholar] [CrossRef]
- Watanabe, J.; Furuya, N.; Fujiwara, Y. Appearance of a BRAF mutation conferring resistance to crizotinib in non-small cell lung cancer harboring oncogenic ROS1 fusion. J. Thorac. Oncol. 2017, 13, e66–e69. [Google Scholar] [CrossRef]
- Ren, S.; Huang, S.; Ye, X.; Feng, L.; Lu, Y.; Zhou, C.; Zhao, J.; He, T.; Wang, J.; Li, B. Crizotinib resistance conferred by BRAF V600E mutation in non–small cell lung cancer harboring an oncogenic ROS1 fusion. Cancer Treat. Res. Commun. 2021, 27, 100377. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, Y.; Chi, X.; Xu, M.; Wang, H. Appearance of an ALK mutation conferring resistance to crizotinib in non–small cell lung cancer harboring oncogenic ROS1 fusion. Lung Cancer 2021, 153, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z.; Han, X.; Li, J.; Guo, H.; Shi, J. Acquired MET D1228N Mutations Mediate Crizotinib Resistance in Lung Adenocarcinoma with ROS1 Fusion: A Case Report. Oncologist 2020, 26, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, P.; Yu, M.; Zhang, Y. Case Report: High-Level MET Amplification as a Resistance Mechanism of ROS1-Tyrosine Kinase Inhibitors in ROS1-Rearranged Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 645224. [Google Scholar] [CrossRef]
- Sudarshan, R.I.; Odintsov, I.; Schoenfeld, A.J.; Siau, E.; Mattar, M.S.; de Stanchina, E.; Khodos, I.; Drilon, A.; Riely, G.J.; Ladanyi, M.; et al. MYC promotes tyrosine kinase inhibitor resistance in ROS1-fusion-positive lung cancer. Mol. Cancer Res. 2022, 20, 722–734. [Google Scholar]
- Priest, K.; Le, A.; Grebregzabheir, A.; Nijmeh, H.; Reis, G.B.; Mandell, M.; Davies, K.D.; Lawrence, C.; O’Donnell, E.; Doebele, R.C.; et al. Evolution of acquired resistance in a ROS1+ KRASG12C+ NSCLC through the MAPK pathway. NPJ. Precis. Oncol. 2023, 7, 9. [Google Scholar] [CrossRef]
- Reischmann, N.; Schmelas, C.; Molina-Vila, M.A.; Jordana-Ariza, N.; Kuntze, D.; Garcia-Roman, S.; Simord, S.A.; Musch, D.; Esdar, C.; Albers, J.; et al. Overcoming MET-mediated resistance in oncogene-driven NSCLC. iScience 2023, 26, 107006. [Google Scholar] [CrossRef]
- Lin, J.J.; Langenbucher, A.; Gupta, P.; Yoda, S.; Fetter, I.J.; Rooney, M.; Do, A.; Kem, M.; Chang, K.P.; Oh, A.Y.; et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ. Precis. Oncol. 2020, 4, 21. [Google Scholar] [CrossRef]
- Wu, C.H.; Su, P.L.; Hsu, C.W.; Chu, C.Y.; Lin, C.C. Small cell transformation in crizotinib-resistant ROS1-rearranged non-small cell lung cancer with retention of ROS1 fusion: A case report. Thorac. Cancer 2021, 12, 3068–3071. [Google Scholar] [CrossRef]
- Dingemans, A.M.; Griesinger, F.; Paz-Ares, L.; Perol, M.; Ren, S.; Hoglander, H.; Kurtsikidze, K.; Siena, S. A randomized phase 3 study of entrectinib versus crizotinib in patients (pts) with locally advanced/metastatic ROS1 fusion-positive (fp) NSCLC with or without baseline CNS metastases (mets). J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS9141. [Google Scholar] [CrossRef]
- Tremblay, G.; Graff, M.; Iadeluca, L.; Daniele, P.; Wilner, K.; Wiltshire, R.; Bartolome, L.; Usari, T.; Cappelleri, J.C.; Camidge, D.R. Effectiveness of crizotinib versus entrectinib in ROS1-positive non-small cell lung cancer using clinical and real-world data. Future Oncol. 2022, 18, 2063–2074. [Google Scholar] [CrossRef] [PubMed]

| Fusion Protein | Frequency in NSCLC (%) | Cellular Location |
|---|---|---|
| CD74-ROS1 (Cluster of differentiation 74) | 49.8 | Endoplasmic reticulum |
| EZR-ROS1 (Ezrin) | 23.6 | Cytoskeleton |
| SDC4-ROS1 (Syndecan) | 9.1 | Endosomes |
| SLC34A2-ROS1 (Solute carrier family 34 member 2) | 5.1 | Endosomes |
| TPM3-ROS1 (Tropomyosin) | 2.9 | Cytoplasm |
| SLC4A4-ROS1 (Solute carrier family 4 member 4) | 1.5 | Endosomes |
| Drug | TRIAL | Patient Number | ORR (with CNS Metastases) | ORR (without CNS Metastases) | DOR at 6 mo (with CNS Metastases) | DOR at 6 mo (without CNS Metastases) |
|---|---|---|---|---|---|---|
| Repotrectinib | TRIDENT-1 | 171 71(TN) 56(1T-CN) 26(1T-1C) 18(2T-CN) | 89(TN) 33(1T-CN) 40(1T-1C) 13(2T-CN) | 76(TN) 41(1T-CN) 44(1T-1C) 40(2T-CN) | 100(TN) 63(1T-CN) 50(1T-1C) 100(2T-CN) | 87(TN) 92(1T-CN) 71(1T-1C) 50(2T-CN) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. ROS1-Rearranged Lung Adenocarcinoma: From Molecular Genetics to Target Therapy. Onco 2023, 3, 189-204. https://doi.org/10.3390/onco3030014
Testa U, Castelli G, Pelosi E. ROS1-Rearranged Lung Adenocarcinoma: From Molecular Genetics to Target Therapy. Onco. 2023; 3(3):189-204. https://doi.org/10.3390/onco3030014
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2023. "ROS1-Rearranged Lung Adenocarcinoma: From Molecular Genetics to Target Therapy" Onco 3, no. 3: 189-204. https://doi.org/10.3390/onco3030014