
Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters


1
Division of Medical Oncology, National Cancer Centre Singapore, Singapore 169610, Singapore
2
Duke-NUS Medical School, National University of Singapore, Singapore 169857, Singapore
Onco 2021, 1(2), 95-100; https://doi.org/10.3390/onco1020008
Submission received: 14 September 2021
/
Revised: 25 October 2021
/
Accepted: 26 October 2021
/
Published: 27 October 2021
(This article belongs to the Special Issue Feature Papers in Onco)
{kind=link}
Abstract
:Simple Summary
The advent of precision oncology has led to growing promise for tumor-agnostic biomarkers, in which molecular biomarkers may select targeted or immunotherapies regardless of the tumor type. Despite this, it remains critical not to disregard the potential importance of the tumor cell of origin. Numerous examples, in which the response to targeted therapies may be markedly influenced by the tumor type, or where there is a predilection for a specific oncogenic driver alterations in a certain tumor type, underlines this. Consequently, an understanding of cell lineage dependency and lineage-survival oncogenes may still offer significant mechanistic insights into disease biology to ultimately identify further therapeutic vulnerabilities.
Abstract
Since the very beginnings of cancer therapy with chemotherapy, tumors have been treated according to the organ or tissue of origin. The advent of precision medicine however, has recently led to growing promise for tumor-agnostic biomarkers for targeted therapies and immunotherapies, such as NTRK fusions. Despite this, prominent examples such as BRAF V600E mutations in melanoma compared to colorectal cancer, in which the site of tumor origin dramatically influences the efficacy of targeted therapies, heeds caution against disregarding the importance of cell of origin. Indeed, another illustrative example, is the almost complete absence outside of cancers originating from the lung of the classical activating EGFR mutations—exon 19 deletions and exon 21 L858R mutations. Consequently, an understanding of lineage dependency and lineage-survival oncogenes may still offer significant mechanistic insights into the malignant transformation of tumors to ultimately identify further therapeutic vulnerabilities.
1. Introduction
Since the very beginnings of cancer therapy with chemotherapy, tumors have been treated according to the organ or tissue of origin [1]. The advent of precision oncology, however, has recently led to growing promise for tumor-agnostic biomarkers for targeted therapies and immunotherapies [2]. Tumor-agnostic or tissue-agnostic biomarkers are molecular signatures or biomarkers used to select therapies regardless of the tumor site of origin [3]. Prominently, NTRK inhibitors entrectinib and larotrectinib are United States (US) Food and Drug Administration (FDA)-approved therapies for patients with advanced solid tumors harboring an NTRK gene fusion. Entrectinib was approved on the basis of an integrated analysis from three multicenter, single-arm, open-label phase 1–2 trials: ALKA, STARTRK-1 and STARTRK-2 [4]. From this pooled subgroup of adult patients with unresectable or metastatic solid tumors with an NTRK gene fusion, there were 54 efficacy-evaluable patients with an objective response rate (ORR) of 57% (95% CI 43.2–70.8) and a median duration of response (DOR) of 10 months (95% CI 7.1 to not estimable). This patient population consisted of ten different tumor types with 19 different histologies, including, most commonly, sarcoma (24%), non-small cell lung cancer (NSCLC; 19%), mammary analogue secretory carcinoma-salivary (13%) and breast cancer (11%). Larotrectinib was similarly approved on the basis of an integrated analysis from three multicenter, single-arm, open-label phase 1–2 trials: LOXO-TRK-14001, SCOUT and NAVIGATE [5]. From 55 patients with solid tumors with an NTRK gene fusion, the ORR was 75% (95% CI 61–85) and the median DOR had not been reached. This initial patient population consisted of 17 tumor types, including, most commonly, salivary gland tumors (22%), other soft tissue sarcomas (20%), infantile fibrosarcoma (13%) and thyroid tumors (9%).
Notably, there are also tumor-agnostic approvals for pembrolizumab. This includes for patients with advanced solid tumors that are microsatellite instability-high or mismatch repair deficient (MSI-H/dMMR), and patients with advanced solid tumors that are tumor mutational burden-high (TMB-H). In patients with advanced MSI-H/dMMR tumors, pembrolizumab has been evaluated in numerous different trials. In KEYNOTE-016, in patients with 12 different tumor types, the ORR was 53% (95% CI 42–64) with no significant difference between colorectal cancer (CRC) and non-CRCs [6]. A larger cohort of MSI-H tumors in KEYNOTE-158 demonstrated an ORR of 34.3% (95% CI 28.3–40.8). This MSI-H cohort consisted of 27 different tumor types, with endometrial (21%), gastric (10.3%), cholangiocarcinoma (9.4%) and pancreatic (9.4%) cancers being most common [7]. In this trial, which comprised other separate non-MSI-H cohorts of less frequently occurring types of solid tumors, a prospective exploratory biomarker analysis was conducted (excluding the aforementioned MSI-H cohort) [8]. There were 102 (13%) out of 790 patients with tissue TMB-H status (assessed as ≥10 mutations per megabase on FoundationOne CDx assay testing), including patients with small cell lung cancer (SCLC; 33%), cervical cancer (16%), endometrial cancer (15%) and anal cancer (14%). The ORR was 30% (95% CI 21–39), with the median duration of response not reached.
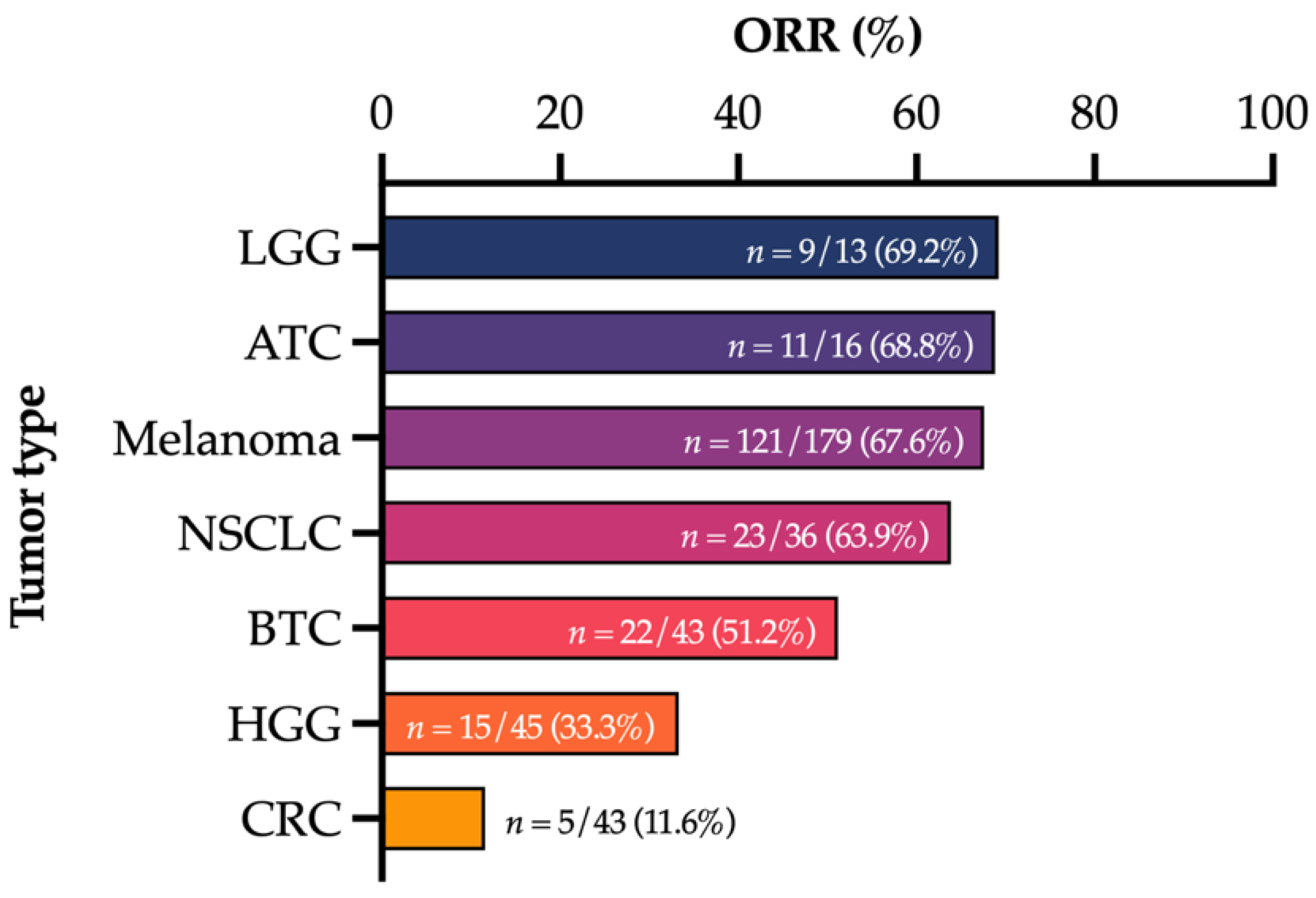
The approval of NTRK inhibitors and pembrolizumab with tissue-agnostic indications has generated significant interest with an increased understanding of the molecular aberrations that may be shared across multiple tumors with distinct sites of origin [9]. This has been driven, in part, by powerful sequencing technologies which allow for the rapid and deep interrogation of tumor samples and generation of large molecular datasets. Consequently, there are increasing numbers of basket trials evaluating therapies across multiple tumor types. For instance, there are promising signs of efficacy for selective RET inhibitors for solid tumors harboring RET alterations [10]. Despite this, prominent examples such as BRAF V600E mutations in melanoma compared to CRC and other tumor types, in which the site of tumor origin dramatically influences the efficacy of targeted therapies [11], heed caution against disregarding the importance of the cell of origin (Figure 1). Whilst BRAF V600E mutations may become targetable in CRC with the addition of an EGFR inhibitor [12], the innate resistance to BRAF inhibitor monotherapy remains fundamentally linked to the tissue of origin. For instance, adaptive feedback signaling networks with reactivation of MAPK signaling are driven by induction of RAS activity from receptor tyrosine kinase (RTK) signaling, particularly EGFR, to a much greater degree, in colorectal cancer compared to melanoma [13,14]. For melanoma, the melanocyte master regulator MITF has also been implicated as a lineage survival oncogene and may cooperate with BRAF V600E for oncogenic transformation [15]. Furthermore, although certain alterations such as NTRK gene fusions may be found across tumor types, there remains a predilection for certain cancers with incidences of >90% in mammary analogue secretory carcinomas (MASC) and secretory breast carcinoma, the reasons for which are not fully understood [16]. Conversely, targetable alterations may be seen in a predominant tumor type. Indeed, the almost complete absence outside of cancers originating from the lung of the classical activating EGFR mutations—exon 19 deletions and exon 21 L858R mutations—is especially illustrative [17]. For immunotherapy biomarkers such as TMB, there is also ongoing debate over the reliability of thresholds across the spectrum of solid tumors to predict response to PD-1 blockade [18,19]. Greater variability in TMB calculation based on NGS targeted panels has also been demonstrated for certain tumor types such as uterine, bladder and CRC compared to lung and head and neck cancers [20]. Our evolving understanding of mutational signatures which may be diverse across tumor types and the subsequent insights into the developmental history of tumors also have important implications for the interpretation of tumor-agnostic biomarkers [21,22]. Therefore, as we strive to optimize therapeutic strategies targeted to the molecular characteristics of an individual patient’s tumor under the premise of precision oncology [23], it is imperative to remain cognizant of the influence of the tumor cell of origin on disease biology.
Data from Subbiah et al. [24], Long et al. [25], Planchard et al. [26], Subbiah et al. [27], Subbiah et al. [28] and Corcoran et al. [29]. ATC—anaplastic thyroid cancer; BTC—biliary tract cancer; CRC—colorectal cancer; HGG—high-grade glioma; LGG—low-grade glioma; NSCLC—non-small cell lung cancer.
Using NSCLC as an example, the cell of origin and genetic driver lesions are increasingly suspected to play a critical role in shaping the phenotypes of lung tumors [30]. Although not definitively proven, adenocarcinomas are thought to arise predominantly from alveolar cells in the distal airways, whilst squamous cell carcinoma and small cell carcinoma arise predominantly from basal cells and neuroendocrine cells in the proximal airways [31]. In animal studies, for example, TP53 inactivation and RB1 loss in neuroendocrine cells have been demonstrated to be sufficient to result in SCLC [32]. The potential for tumor lineage plasticity, however, has also been demonstrated, with loss of TTF-1/NKX2-1 implicated in the development of either mucinous adenocarcinoma with concurrent oncogenic KRAS mutation or squamous cell carcinoma with SOX2 gain [33]. Lineage-defining transcription factors may also shape the tumor immune microenvironment [34]. In EGFR-mutated NSCLC, early descriptions of EGFR mutations in lung adenocarcinoma identified strong correlations with immunohistochemical expression of TTF-1 (also known as NKX2-1) and anatomical terminal respiratory unit (TRU) histomorphology [35]. Importantly, TTF-1/NKX2-1 expression is used in routine clinical practice as a sensitive marker for differentiating lung adenocarcinoma from other lung cancer histologies [36]. In fact, EGFR and TTF-1/NKX2-1 have both been shown to be strong oncogenic drivers, sufficient to transform a pre-invasive lesion into an invasive adenocarcinoma, potentially without concurrent driver genomic alterations [37]. However, TTF-1/NKX2-1 is also a lung lineage master regulator gene, critical in lung morphogenesis and embryological development, and differentiation of distal pulmonary alveolar cells [38]. Taken together, this builds on evidence that cell lineage-specific pathways and the corresponding transcription factors that determine pulmonary epithelial differentiation may have a key role in different histologic and molecular subtypes of lung cancer [39]. In The Cancer Genome Atlas (TCGA) description of lung adenocarcinoma, validated transcriptional molecular subtypes consisting of terminal respiratory unit (TRU), proximal inflammatory (PI) and proximal proliferative (PP) were each shown to enrich for certain oncogenic driver mutations or translocations along with other genomic features [40]. The transcriptional TRU subtype harbored the majority of EGFR-mutated tumors. Notably, these transcriptional molecular subtypes were originally identified from correlations with histopathological and anatomical descriptions of lung tumors [41]. Finally, histological transformation with small cell or squamous transformation as a commonly identified mechanism of resistance to the selective pressure induced by EGFR inhibition further highlights the importance of cell lineage [42]. Therefore, oncogenic dependency, phenotypic plasticity and subsequent response to therapy may all be influenced by the underlying cell of origin [43].
2. Conclusions
Despite the promise of tumor-agnostic biomarkers for precision oncology, it remains critical to not disregard the potential importance of the cell of origin. An understanding of lineage dependency and lineage survival oncogenes may still offer significant mechanistic insights into disease biology. Ultimately, this may identify further therapeutic vulnerabilities whilst remaining within the umbrella of precision oncology.
Funding
This research received no external funding.
Conflicts of Interest
A.C.T. reports consultant or advisory roles for Amgen outside the submitted work.
References
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Photopoulos, J. The Future of Tissue-Agnostic Drugs. Nature 2020, 585, S16–S18. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 184–187. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1-2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021, 7, 15–28. [Google Scholar] [CrossRef]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A Phase 1 Study of LOXO-292, a Potent and Highly Selective RET Inhibitor, in Patients with RET-Altered Cancers. J. Clin. Oncol. 2018, 36, 102. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Re-Activation of MAPK Signaling Contributes to Insensitivity of BRAF Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of Colon Cancer to BRAF(V600E) Inhibition through Feedback Activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative Genomic Analyses Identify MITF as a Lineage Survival Oncogene Amplified in Malignant Melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Absence of EGFR Mutation in the Kinase Domain in Common Human Cancers besides Non-Small Cell Lung Cancer. Int. J. Cancer 2005, 113, 510–511. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High Tumor Mutation Burden Fails to Predict Immune Checkpoint Blockade Response across All Cancer Types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.-J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing Guidelines to Harmonize Tumor Mutational Burden (TMB): In Silico Assessment of Variation in TMB Quantification across Diagnostic Platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Seoane, J.; Le Tourneau, C.; Siu, L.L.; Marais, R.; Michiels, S.; Soria, J.C.; Campbell, P.; Normanno, N.; Scarpa, A.; et al. The European Society for Medical Oncology (ESMO) Precision Medicine Glossary. Ann. Oncol. 2018, 29, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus Trametinib in Patients with Previously Untreated BRAFV600E-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-Mutated Biliary Tract Cancer (ROAR): A Phase 2, Open-Label, Single-Arm, Multicentre Basket Trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Subbiah, V.; Stein, A.; van den Bent, M.; Wick, A.; de Vos, F.Y.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; Campone, M.; et al. Abstract CT025: Dabrafenib plus Trametinib in BRAF V600E-Mutant High-Grade (HGG) and Low-Grade Glioma (LGG). Cancer Res. 2021, 81, CT025. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of Origin of Lung Cancers: Lessons from Mouse Studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Govindan, R. Clinical Implications of Genomic Discoveries in Lung Cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, P.R.; Chow, R.D.; Saladi, S.V.; Tata, A.; Konkimalla, A.; Bara, A.; Montoro, D.; Hariri, L.P.; Shih, A.R.; Mino-Kenudson, M.; et al. Developmental History Provides a Roadmap for the Emergence of Tumor Plasticity. Dev. Cell 2018, 44, 679–693.e5. [Google Scholar] [CrossRef] [Green Version]
- Mollaoglu, G.; Jones, A.; Wait, S.J.; Mukhopadhyay, A.; Jeong, S.; Arya, R.; Camolotto, S.A.; Mosbruger, T.L.; Stubben, C.J.; Conley, C.J.; et al. The Lineage-Defining Transcription Factors SOX2 and NKX2-1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity 2018, 49, 764–779.e9. [Google Scholar] [CrossRef] [Green Version]
- Yatabe, Y.; Kosaka, T.; Takahashi, T.; Mitsudomi, T. EGFR Mutation Is Specific for Terminal Respiratory Unit Type Adenocarcinoma. Am. J. Surg. Pathol. 2005, 29, 633–639. [Google Scholar] [CrossRef]
- Yatabe, Y.; Dacic, S.; Borczuk, A.C.; Warth, A.; Russell, P.A.; Lantuejoul, S.; Beasley, M.B.; Thunnissen, E.; Pelosi, G.; Rekhtman, N.; et al. Best Practices Recommendations for Diagnostic Immunohistochemistry in Lung Cancer. J. Thorac. Oncol. 2019, 14, 377–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamura, K. Clinicopathological Characteristics and Mutations Driving Development of Early Lung Adenocarcinoma: Tumor Initiation and Progression. Int. J. Mol. Sci. 2018, 19, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herriges, M.; Morrisey, E.E. Lung Development: Orchestrating the Generation and Regeneration of a Complex Organ. Development 2014, 141, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.K.C.; Nguyen, D.X. Lineage Factors and Differentiation States in Lung Cancer Progression. Oncogene 2015, 34, 5771–5780. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [PubMed]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [Green Version]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of Acquired Resistance to First- and Second-Generation EGFR Tyrosine Kinase Inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef]
- Garraway, L.A.; Sellers, W.R. Lineage Dependency and Lineage-Survival Oncogenes in Human Cancer. Nat. Rev. Cancer 2006, 6, 593–602. [Google Scholar] [CrossRef]
Figure 1.
Spectrum of response rates to combination BRAF and MEK inhibition with dabrafenib and trametinib across a range of BRAF V600E-mutated tumor types.
Figure 1.
Spectrum of response rates to combination BRAF and MEK inhibition with dabrafenib and trametinib across a range of BRAF V600E-mutated tumor types.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, A.C. Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters. Onco 2021, 1, 95-100. https://doi.org/10.3390/onco1020008
AMA Style
Tan AC. Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters. Onco. 2021; 1(2):95-100. https://doi.org/10.3390/onco1020008
Chicago/Turabian StyleTan, Aaron C. 2021. "Tumor-Agnostic Biomarkers: Heed Caution, and Why Cell of Origin Still Matters" Onco 1, no. 2: 95-100. https://doi.org/10.3390/onco1020008