

Limitations of Protein Structure Prediction Algorithms in Therapeutic Protein Development



Abstract
:Simple Summary
Abstract

1. Introduction
2. Finding Applications
3. Testing a New Application
4. Materials and Methods
4.1. Data Collection
4.2. Structure Prediction and Scores
4.3. Physiochemical Properties
4.4. Protein Interactions
5. Results
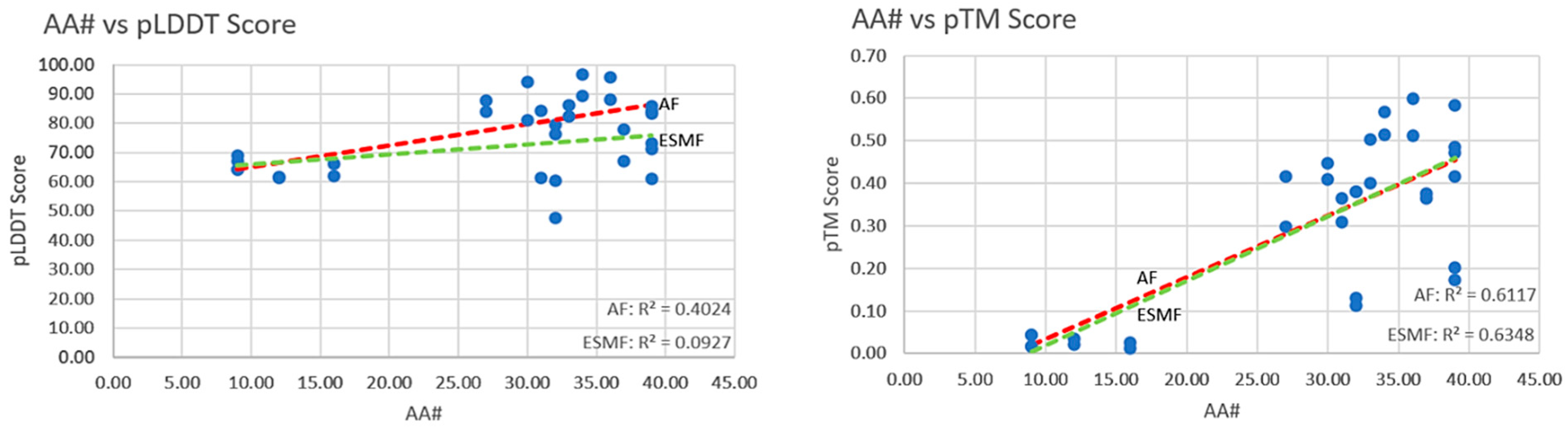
5.1. Orthogonal Comparison—AF2 vs. ESMF
5.2. Complexity of Structures and Prediction Scores
5.3. Physiochemical Attributes and 3D Structure
5.4. Protein Interactions—Effects of Structural Folds
6. Discussion
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cyrus, L. How to Fold Graciously. In Mossbauer Spectroscopy in Biological Systems: Proceedings of a Meeting Held at Allerton House; University of Illinois Bulletin: Monticello, IL, USA, 1969; pp. 22–24. [Google Scholar]
- Hirata, F.; Sugita, M.; Yoshida, M.; Akasaka, K. Perspective: Structural fluctuation of protein and Anfinsen’s thermodynamic hypothesis. J. Chem. Phys. 2018, 148, 020901. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.; Li, Y.; Omenn, G.S.; Zhang, Y. Fast and accurate Ab Initio Protein structure prediction using deep learning potentials. PLoS Comput. Biol. 2022, 18, e1010539. [Google Scholar] [CrossRef]
- Corey, R.B.; Kuntz, I.D. ENCEPP: A program for predicting the conformational geometry of organic molecules. J. Comput. Chem. 1974, 2, 287–303. [Google Scholar]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins 2021, 89, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Zitnick, C.L. Meta’s Genomics AI ESMFold Predicts Protein Structure 6x Faster Than AlphaFold2. InfoQ. 2022. Available online: https://www.infoq.com/news/2022/08/meta-genomic-ai-esmfold/ (accessed on 11 May 2023).
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.G.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Peng, J.; Xu, J. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins Struct. Funct. Bioinform. 2011, 79 (Suppl. 10), 161–171. [Google Scholar] [CrossRef]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novos tructure prediction from primary sequence. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins Struct. Funct. Bioinform. 2008, 77 (Suppl. 9), 100–113. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Schwede, T. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak RBradley, P. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Montanucci, L.; Capriotti, E.; Frank, Y.; Ben-Tal, N.; Fariselli, P. DDGun: An untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinform. 2019, 20, S14. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef]
- Lv, X.; Chen, J.; Lu, Y.; Chen, Z.; Xiao, N.; Yang, Y. Accurately Predicting Mutation-Caused Stability Changes from Protein Sequences Using Extreme Gradient Boosting. J. Chem. Inf. Model. 2020, 60, 2388–2395. [Google Scholar] [CrossRef]
- Yin, J.; Lei, J.; Yu, J.; Cui, W.; Satz, A.L.; Zhou, Y.; Feng, H.; Deng, J.; Su, W.; Kuai, L. Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target. Molecules 2022, 27, 5797. [Google Scholar] [CrossRef]
- Gao, M.; An, D.N.; Parks, J.M.; Skolnick, J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat. Commun. 2022, 13, 1744. [Google Scholar] [CrossRef]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for Protein Complex Modeling Reveals Accuracy Determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef]
- Available online: https://pubmed.ncbi.nlm.nih.gov/?term=alphafold (accessed on 11 May 2023).
- Available online: https://pubmed.ncbi.nlm.nih.gov/?term=ESMFold (accessed on 11 May 2023).
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The protein data bank archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [PubMed]
- Dana, J.M.; Gutmanas, A.; Tyagi, N.; Qi, G.; O’donovan, C.; Martin, M.; Velankar, S. SIFTS: Updated structure integration with function, taxonomy and sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019, 47, D482–D489. [Google Scholar] [CrossRef] [PubMed]
- Mosalaganti, S.; Obarska-Kosinska, A.; Siggel, M.; Taniguchi, R.; Turoňová, B.; Zimmerli, C.E.; Buczak, K.; Schmidt, F.H.; Margiotta, E.; Mackmull, M.-T.; et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 2022, 376, eabm9506. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, S.; Ser, Z.; Kuang, H.; Than, T.; Guan, D.; Zhao, X.; Patel, D.J. Cryo-EM structure of DNA-bound Smc5/6 reveals DNA clamping enabled by multi-subunit conformational changes. Proc. Natl. Acad. Sci. USA 2022, 119, e2202799119. [Google Scholar] [CrossRef]
- Hötzel, I. Deep-Time Structural Evolution of Retroviral and Filoviral Surface Envelope Proteins. J. Virol. 2022, 96, e0006322. [Google Scholar] [CrossRef]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Caldararu, O.; Blundell, T.L.; Kepp, K.P. A base measure of precision for protein stability predictors: Structural sensitivity. BMC Bioinform. 2021, 22, 88. [Google Scholar] [CrossRef]
- Goulet, A.; Cambillau, C. Present Impact of AlphaFold2 Revolution on Structural Biology, and an Illustration with the Structure Prediction of the Bacteriophage J-1 Host Adhesion Device. Front. Mol. Biosci. 2022, 9, 907452. [Google Scholar] [CrossRef]
- Anbo, H.; Sakuma, K.; Fukuchi, S.; Ota, M. How AlphaFold2 Predicts Conditionally Folding Regions Annotated in an Intrinsically Disordered Protein Database, IDEAL. Biology 2023, 12, 182. [Google Scholar] [CrossRef]
- Saldaño, T.; Escobedo, N.; Marchetti, J.; Zea, D.J.; Mac Donagh, J.; Velez Rueda, A.J.; Gonik, E.; García Melani, A.; Novomisky Nechcoff, J.; Salas, M.N.; et al. Impact of protein conformational diversity on AlphaFold predictions. Bioinformatics 2022, 38, 2742–2748. [Google Scholar] [CrossRef]
- Roney, J.P.; Ovchinnikov, S. State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold. Phys. Rev. Lett. 2022, 129, 238101. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Perez, A. AlphaFold encodes the principles to identify high affinity peptide binders. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Chakravarty, D.; Porter, L.L. AlphaFold2 fails to predict protein fold switching. Protein Sci. 2022, 31, e4353. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://alphafold.ebi.ac.uk/faq (accessed on 11 May 2023).
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://torchmetrics.readthedocs.io/en/stable/classification/auroc.html (accessed on 11 May 2023).
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; SRaghava, G.P. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [PubMed]
- FDA Purplebook. (n.d.-b). Available online: https://purplebooksearch.fda.gov/ (accessed on 28 January 2023).
- Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations. (n.d.); FDA: Rockville, MD, USA, 2023. Available online: https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm (accessed on 28 January 2023).
- Available online: https://webs.iiitd.edu.in/raghava/thpdb/length.php (accessed on 11 May 2023).
- NCATS Inxight Drugs. (n.d.). Available online: https://drugs.ncats.io/ (accessed on 11 May 2023).
- KEGG Pathways Database. Available online: https://www.genome.jp/kegg/pathway.html (accessed on 11 May 2023).
- DrugBank Online|Database for Drug and Drug Target Info. (n.d.). DrugBank. Available online: https://go.drugbank.com/ (accessed on 11 May 2023).
- FDA. ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin. Available online: https://www.fda.gov/media/107622/download (accessed on 10 July 2023).
- Available online: https://www.cusabio.com/m-299.html#a03 (accessed on 11 May 2023).
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Google Colaboratory. (n.d.). Available online: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/ESMFold.ipynb#scrollTo=CcyNpAvhTX6q (accessed on 11 May 2023).
- Expasy-ProtParam tool. (n.d.). Available online: https://web.expasy.org/protparam/ (accessed on 15 April 2023).
- Structural Characterization Methods for Biosimilars: Fit-for-Purpose, Qualified or Validated-GaBI Journal. (n.d.). Available online: http://gabi-journal.net/structural-characterization-methods-for-biosimilars-fit-for-purpose-qualified-or-validated.html (accessed on 11 May 2023).
- LZerD Web Server. (n.d.). Available online: https://lzerd.kiharalab.org/ (accessed on 11 May 2023).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. GOAP: A generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys. J. 2011, 101, 2043–2052. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar]
- Huang, S.Y.; Zou, X. ITScorePro: An efficient scoring program for evaluating the energy scores of protein structures for structure prediction. Protein Struct. Predict. 2014, 71–81. [Google Scholar]
- Prodigy Webserver. (n.d.). Available online: https://wenmr.science.uu.nl/prodigy/ (accessed on 11 May 2023).
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, I. AlphaFold Distillation for Improved Inverse Protein Folding. OpenReview. Available online: https://openreview.net/forum?id=brk7Ct4Tb1M (accessed on 29 September 2022).
- Campo, D.S.; Dimitrova, Z.; Khudyakov, Y. Physicochemical Correlation between Amino Acid Sites in Short Sequences under Selective Pressure. In Proceedings of the Bioinformatics Research and Applications: Fourth International Symposium, ISBRA 2008, Atlanta, GA, USA, 6–9 May 2008; pp. 146–158. [Google Scholar]
- He, Y.; Rackovsky, S.; Yin, Y.; Scheraga, H.A. Alternative approach to protein structure prediction based on sequential similarity of physical properties. Proc. Natl. Acad. Sci. USA 2015, 112, 5029–5032. [Google Scholar] [CrossRef] [PubMed]
- Pok, G.; Jin, C.; Ryu, K.H. Correlation of Amino Acid Physicochemical Properties with Protein Secondary Structure Conformation. In Proceedings of the 2008 International Conference on BioMedical Engineering and Informatics, Sanya, China, 27–30 May 2008. [Google Scholar]
- Saghapour, E.; Sehhati, M. Physicochemical Position-Dependent Properties in the Protein Secondary Structures. Iran. Biomed. J. 2019, 23, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Nupur, N.; Joshi, S.; Gulliarme, D.; Rathore, A.S. Analytical Similarity Assessment of Biosimilars: Global Regulatory Landscape, Recent Studies and Major Advancements in Orthogonal Platforms. Front. Bioeng. Biotechnol. 2022, 10, 832059. [Google Scholar]
- Rigi, G.; Kardar, G.; Hajizade, A.; Zamani, J.; Ahmadian, G. The effects of a truncated form of Staphylococcus aureus protein A (SpA) on the expression of cytokines of autoimmune patients and healthy individuals. Res. Sq. 2022. [Google Scholar]
- Stevens, A.O.; He, Y. Benchmarking the Accuracy of AlphaFold 2 in Loop Structure Prediction. Biomolecules 2022, 12, 985. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.-P.; Gardella, T.J. PTH receptor-1 signalling—mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar]
- Kastritis, P.L.; Rodrigues, J.D.; Folkers, G.E.; Boelens, R.; Bonvin AM, J.J. Proteins Feel More Than They See: Fine-Tuning of Binding Affinity by Properties of the Non-Interacting Surface. J. Mol. Biol. 2014, 426, 2632–2652. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Yokota, K.; Fukui, K. Energy based approach for understanding the recognition mechanism in protein–protein complexes. Mol. Biosyst. 2009, 5, 1779–1786. [Google Scholar] [CrossRef]
- Hilario, E.C.; Stern, A.; Wang, C.H.; Vargas, Y.W.; Morgan, C.J.; Swartz, T.E.; Patapoff, T.W. An Improved Method of Predicting Extinction Coefficients for the Determination of Protein Concentration. PDA J. Pharm. Sci. Technol. 2017, 71, 127–135. [Google Scholar] [CrossRef]
- Yuan, H.; Li, Z.; Wang, X.; Qi, R. Photodynamic Antimicrobial Therapy Based on Conjugated Polymers. Polymers 2022, 14, 3657. [Google Scholar] [CrossRef] [PubMed]
- Manhart, M.; Morozov, A.V. Protein folding and binding can emerge as evolutionary spandrels through structural coupling. Proc. Natl. Acad. Sci. USA 2015, 112, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. The role of charged surface residues in the binding ability of small hubs in protein-protein interaction networks. Biophysics 2007, 3, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, W.; Li, W.; Zhen, X.; Liang, J.; Cai, W.; Xu, F.; Yuan, K.; Lin, G.N. Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction. Biology 2022, 11, 1454. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Chen, Z.; Zhang, C.; Xie, Y.; Ovchinnikov, S.G.; Gao, Y.Q.; Liu, S. ColabDock: Inverting AlphaFold structure prediction model for protein-protein docking with experimental restraints. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Scardino, V.; Di Filippo, J.I.; Cavasotto, C.N. How good are AlphaFold models for docking-based virtual screening? iScience 2022, 26, 105920. [Google Scholar] [CrossRef]
- Johansson-Åkhe, I.; Wallner, B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front. Bioinform. 2022, 2, 85. [Google Scholar] [CrossRef]
- Tang, Q.; Ren, W.; Wang, J.; Kaneko, K. The Statistical Trends of Protein Evolution: A Lesson from AlphaFold Database. Mol. Biol. Evol. 2022, 39, msac197. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–625. [Google Scholar] [CrossRef]
- Available online: https://yanglab.nankai.edu.cn/trRosetta/ (accessed on 12 August 2023).
- Available online: https://predictioncenter.org/casp15/zscores_final.cgi (accessed on 12 August 2023).
- Random Sequence Generator-Random DNA, RNA or Protein Sequences. (n.d.). Available online: https://molbiotools.com/randomsequencegenerator.php (accessed on 29 April 2023).
- Thomas, J.; Ramakrishnan, N.; Bailey-Kellogg, C. Graphical models of residue coupling in protein families. In Proceedings of the 5th International Workshop on Bioinformatics, Chicago, IL, USA, 7 May 2008; pp. 12–20. [Google Scholar]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green THassabis, D. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. End-to-End Differentiable Learning of Protein Structure. Cell Syst. 2019, 8, 292–301.e3. [Google Scholar] [CrossRef] [PubMed]
- Ismi, D.P.; Pulungan, R. Deep learning for protein secondary structure prediction: Pre and post-AlphaFold. Comput. Struct. Biotechnol. J. 2022, 20, 6271–6286. [Google Scholar] [CrossRef] [PubMed]
- Godzik, A. Metagenomics and the protein universe. Curr. Opin. Struct. Biol. 2011, 21, 398–403. [Google Scholar] [CrossRef]
- Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [CrossRef]
- Laurents, D.V. AlphaFold 2 and NMR Spectroscopy: Partners to understand protein structure, dynamics and function. Front. Mol. Biosci. 2022, 9, 906437. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| PTH Residue | PTH Residue Number | AF2 Residual pLDDT | ESMF Residual pLDDT (Average) |
|---|---|---|---|
| Ser | 1 | 85.82 | 48.86 |
| Ser | 3 | 94.47 | 66.06 |
| Glu | 4 | 95.84 | 63.25 |
| Ile | 5 | 96.16 | 65.74 |
| Leu | 7 | 96.63 | 65.94 |
| Met | 8 | 97.32 | 68.10 |
| Leu | 11 | 97.24 | 61.35 |
| His | 14 | 96.93 | 64.82 |
| Leu | 15 | 97.02 | 69.84 |
| Ser | 17 | 96.32 | 66.47 |
| Met | 18 | 96.94 | 66.27 |
| Glu | 19 | 96.60 | 48.86 |
| Arg | 20 | 96.58 | 66.06 |
| Phe | 34 | 97.32 | 63.25 |
| Query Coverage (%) | Percentage Identity (%) | AF pLDDT | AF pTM | ESMF pLDDT | ESMF pTM | |
|---|---|---|---|---|---|---|
| Trastuzumab: | ||||||
| original | 99.00 | 93.73 | 91.00 | 0.61 | 82.01 | 0.58 |
| one-domain-mutated | 99.00 | 75.43 | 79.50 | 0.53 | 71.90 | 0.46 |
| all-domains-mutated | 3.00 | 100.00 | 25.20 | 0.15 | 19.19 | 0.13 |
| Etanercept: | ||||||
| original | 49.00 | 100.00 | 82.10 | 0.47 | 79.23 | 0.41 |
| one-domain-mutated | 37.00 | 100.00 | 68.50 | 0.38 | 68.34 | 0.39 |
| all-domains-mutated | 0.00 | 0.00 | 32.20 | 0.17 | 24.84 | 0.13 |
| Coagulation Factor-VIIa: | ||||||
| original | 62.00 | 100.00 | 86.10 | 0.77 | 87.42 | 0.79 |
| one-domain-mutated | 37.00 | 100.00 | 48.80 | 0.25 | 43.46 | 0.24 |
| all-domains-mutated | 25.00 | 40.87 | 28.10 | 0.18 | 25.19 | 0.14 |
| Darbepoetin alfa: | ||||||
| original | 86.00 | 95.18 | 87.70 | 0.84 | 83.95 | 0.85 |
| domain-mutated | 0.00 | 0.00 | 40.00 | 0.29 | 41.64 | 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Mariam, Z.; Paracha, R.Z. Limitations of Protein Structure Prediction Algorithms in Therapeutic Protein Development. BioMedInformatics 2024, 4, 98-112. https://doi.org/10.3390/biomedinformatics4010007
Niazi SK, Mariam Z, Paracha RZ. Limitations of Protein Structure Prediction Algorithms in Therapeutic Protein Development. BioMedInformatics. 2024; 4(1):98-112. https://doi.org/10.3390/biomedinformatics4010007
Chicago/Turabian StyleNiazi, Sarfaraz K., Zamara Mariam, and Rehan Z. Paracha. 2024. "Limitations of Protein Structure Prediction Algorithms in Therapeutic Protein Development" BioMedInformatics 4, no. 1: 98-112. https://doi.org/10.3390/biomedinformatics4010007