1. Introduction
State-of-the-art machine learning (ML)-based artificial intelligence (AI) algorithms are able to effectively and efficiently diagnose (classify) high-dimensional datasets in modern medicine; see [
1] for an overview. In particular, for multiparameter flow cytometry data, see [
2,
3]. These systems use one set of data (learning data) to develop (train/learn) an algorithm that is able to classify data that are not part of the training data (i.e., the testing or validation data). This strategy is called supervised learning [
4]. The most successful supervised learning methods are, among others, like specific neural networks [
5], the family of random forest classifiers [
6]. Within AI, these algorithms are called subsymbolic classifiers [
7]. Subsymbolic systems are able to perform a task (skill), such as assigning the most suitable diagnosis to a case. However, it is meaningless and impossible to ask a subsymbolic AI system for an explanation or reason for its decisions (e.g., [
8]). To circumvent this lack of explainability, there has been a recent explosion of work in which a second (post hoc) model (“post hoc explainer”) is created to explain a black-box ML model [
9]. Alternatively, psychological research has proposed category norms derived from recruiting exemplars as representations of knowledge [
10]. In ML, this psychological concept is called prototyping [
11] and is well-known in cluster analysis (e.g., [
12]) and neural network studies (e.g., [
13,
14]). Recently, Angelov and Soares proposed integrating prototype selection into a deep-learning network [
15]. A pretrained traditional deep-learning-based image classifier (a convolutional neural network (CNN)) was combined with a prototype selection process to form an “explainable” system [
15].
In particular, in medicine, explanations and reasons for the decisions made by algorithms concerning the health states or treatment options of patients are required by law, e.g., the General Data Protection Regulation (GDPR) in the EU [
16] (European Parliament and Council: General Data Protection Regulation (GDPR), in effect since 25 May 2018). This calls for systems that produce human-understandable knowledge out of the input data and base their decisions on this knowledge so that these systems can explain the reasons for particular decisions. Such systems are called “symbolic” or “explainable AI (XAI)” [
17] systems. If such systems aim to represent their reasoning in a form that is understandable to application domain users, they are called “knowledge-based” or “expert” systems [
18].
Some XAI systems produce classification rules from a set of conditions. If the conditions are fulfilled, a particular diagnosis is derived. The conditions of the rules consist of logical statements regarding the parameter (variable) ranges. For example, a “thrombocyte” can be described with the rule “CD45- and CD42+”. The term “CD45-” denotes the condition that the expression of CD45 structures is low on a cell’s surface. In symbolic AI systems, the production of diagnostic rules from datasets is a classic approach. Algorithms such as classification and regression trees (CART) [
19], C4.5 [
20], and RIPPER [
21] have already been developed in the last century. However, these algorithms aim to optimize their classification performance and not to achieve the best understandability of their rules for domain users.
One of the essential requirements for the human understandability of machine-generated knowledge is simplicity. If a rule comprises too many conditions or if there are too many rules, their meanings are very hard or impossible to understand. In AI, description simplicity is a standard quality measure for understandability [
22]. Therefore, we use here a well-known practical measure for optimal simplicity [
23], see below.
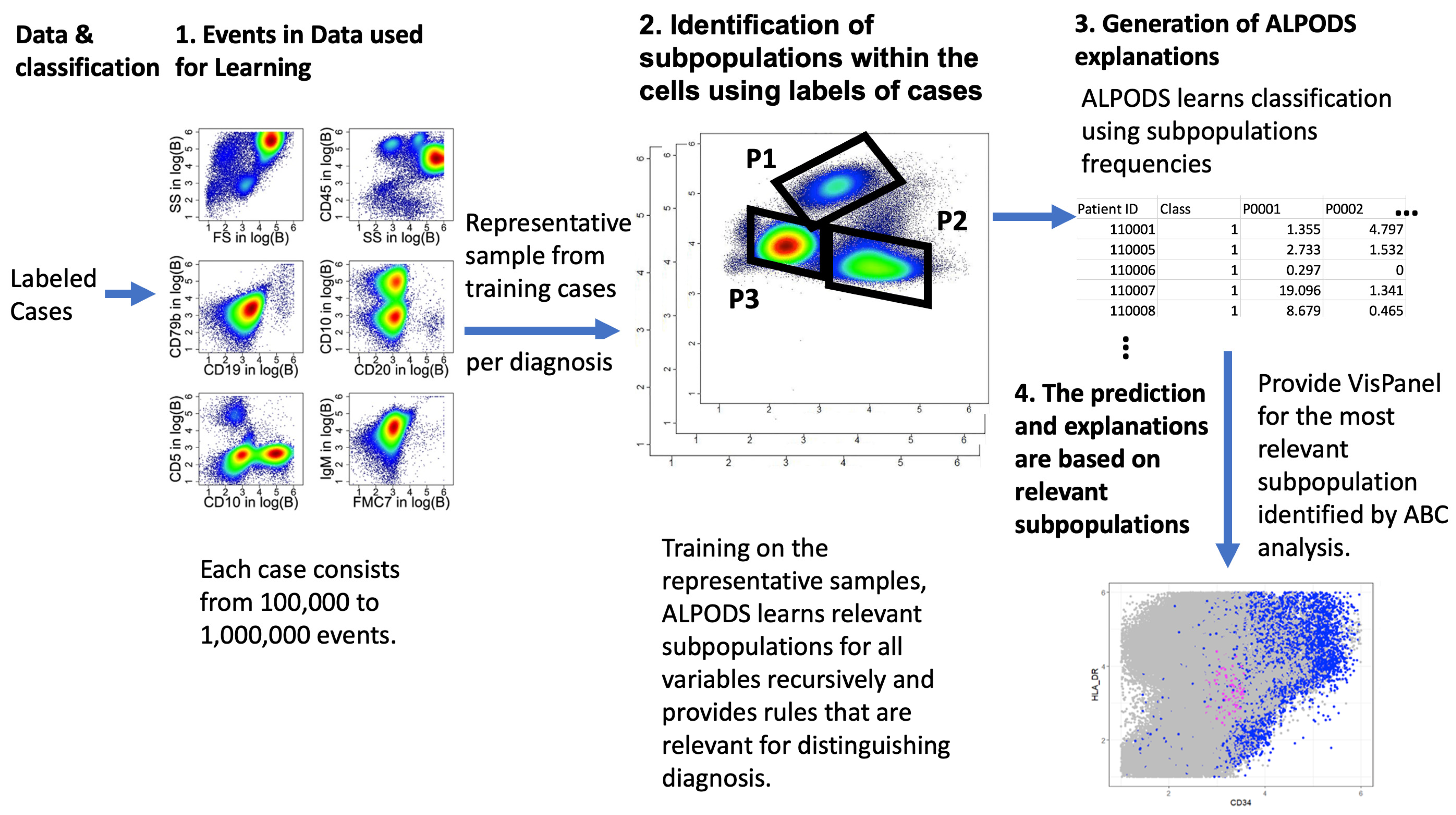
The objective of the study is to address the challenge faced by artificial intelligence (AI) systems in simultaneously diagnosing large data with high accuracy and proving comprehensible rules. Such large datasets typically involve analyzing 10 to 30 features of over 100,000 events for a high number of patients.
This work proposes a symbolic ML algorithm (algorithmic population descriptions (ALPODS)) that produces and utilizes user-understandable knowledge for its decisions. The algorithm is tested on a typical ML example and two multiparameter flow cytometry datasets from routine clinical practice. The latter datasets are acquired from two clinical centers (Marburg and Dresden). The problem at hand is deciding the primary origin of a probe: mostly bone marrow (BM) or peripheral blood (PB).
Our key contributions are as follows:
Algorithmic population descriptions (ALPODS) is a novel supervised explainable AI (XAI) that provides physicians with tailored explanations in biological datasets.
ALPODS is fast-working and requires for learning only a very low number of cases.
ALPODS distinguishes normal controls from leukemia and bone marrow from peripheral blood samples with high accuracy.
ALPODS outperforms comparable systems on several datasets in terms of interpretability, performance, and processing time.
The XAI was already successfully applied for the hemodilution in BM samples to prevent false negative MRD reports and was able to identify to physicians prior unknown and highly predictive cell populations and improved chronic lymphocytic leukemia outcome predictions.
The proposed algorithm is compared to rule-generating AI systems [
24] and decision tree rules [
25], as well as recently published rule-generating algorithms for flow cytometry [
26,
27] which are introduced in the
Section 2.
5. Results
The algorithms introduced in the prior sections were applied to the synthetic data with Gaussian noise derived from Iris. This serves as a basic test of their performance because the datasets for human medical research are limited and not available in large quantities. In contrast, Iris is a well-known biological dataset for which the performance of algorithms is expected to be high. The data comprised three distinct classes (
), which needed to be diagnosed using the
variables.
Table 1 shows that the accuracies of all algorithms except SuperFlowType exceeded
. The largest differences were observed in the number of identified partitions and the number of conditions required for the description of a partition. However, all algorithms delivered sufficiently simple descriptions with typically fewer than nine partitions. Surprisingly, the partitions delivered by FAUST did not match the partitions of the synthetic data, meaning that the descriptions were not useful.
The eUD3.5 algorithm could not be transferred to the Marburg and Dresden datasets because no source code was accessible. For each method with usable open-source code, subpopulation rules were constructed on the two training datasets (for details, see
Section 3). The physicians required that computational time per method and experiment should be limited to
72 h. The accuracies achieved by the resulting classifiers on the test data were calculated on the respective sets of patient data files using up to
50 rounds of cross-validation within the time limit. Neither RF_LIME nor SuperFlowType were able to compute results on a personal computer. Nevertheless, neither the RF nor FlowType submethod (of RF_LIME and SuperFlowType, respectively) provided full results on the training set. Thus, a
sample was used, which resulted in a
40 h computation time for RF_LIME and a
h computation time for SuperFlowType in the case of the Marburg dataset.
Table 2 shows these results.
For the Dresden dataset, RF_LIME did not obtain a result in the required maximum duration of the experiment. Computing the results for SuperFlowType took 24 h, again with the procedure defined above.
Table 3 presents these results. Contrary to the compared algorithms, ALPODS finished after 1 min of CPU time on the iMac PRO.Therefore, only ALPODS was able to perform the desired 50 cross-validations. For the Marburg dataset, ALPODS identified five relevant populations for distinguishing between BM and PB. Utilizing visualization techniques, clinical experts could understand the population and identify the following cell types in the population.
FlowType started with all possible conditions for all d variables. For the flow cytometric data, this meant more than populations. Optimix reduced this to approximately populations, and the subsequent ABC analysis, which took the significance of the subpopulations into account for diagnosis purposes, reduced this to an average of populations. However, this number of subpopulations was too large to even look for further explanations. Only a fraction of the results had acceptable accuracies exceeding The number of subpopulations identified by RF_LIME typically ranged from to .
Contrary to the claim of the authors of FAUST that “it has a general purpose and can be applied to any collection of related real-valued matrices one wishes to partition” [
32], FAUST failed to reproduce the cluster structures of the synthetic dataset derived from iris (0.64 accuracy). As their initial benchmark was neither unbiased nor compared with state-of-the-art clustering algorithms (cf. the discussion in [
72]), further studies would be required to investigate whether FAUST is able to explain the given structures in data. Although the RF classifier yielded a performance nearly equal to that of ALPODS for the flow cytometry datasets, a large number of populations with eight conditions for each population were too complex to be understandable by domain experts without additional algorithms for selecting appropriate populations. Even then, describing each population by eight conditions for each condition containing up to three thresholds seems unfeasible. The results indicate that FAUST is probably not very useful for discovering knowledge in flow cytometric datasets. Moreover, its computational time increased on larger training datasets in comparison with ALPODS.
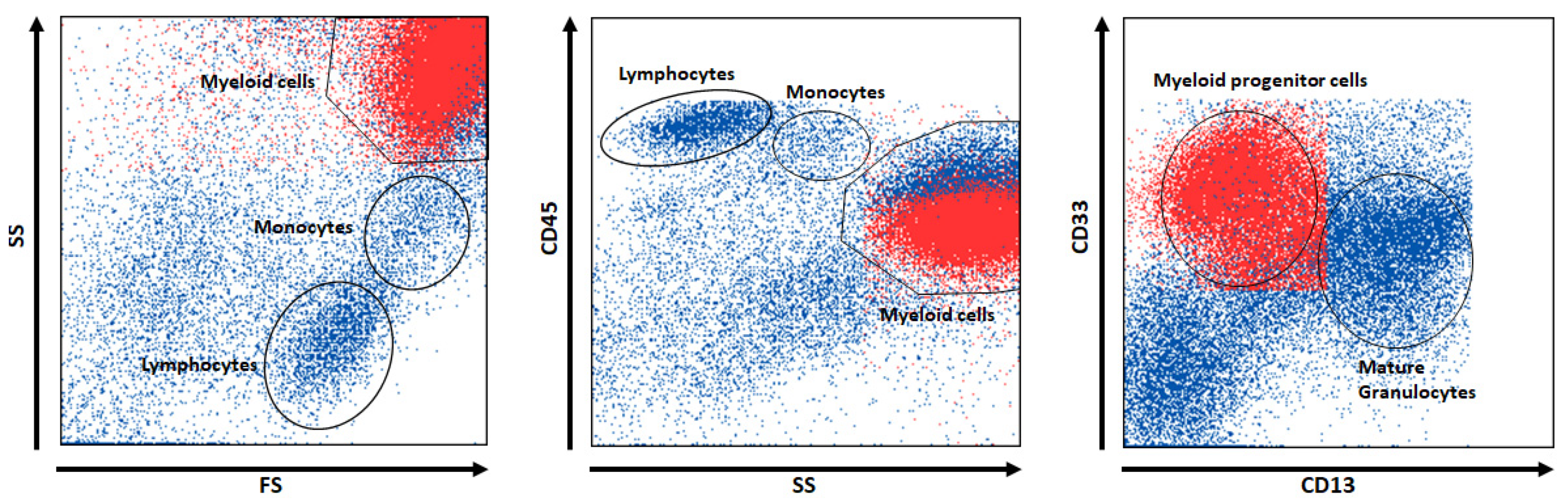
The results obtained on the Dresden dataset were similar. For the five populations identified by ALPODS (see
Table 4), the main cell types could be identified (see
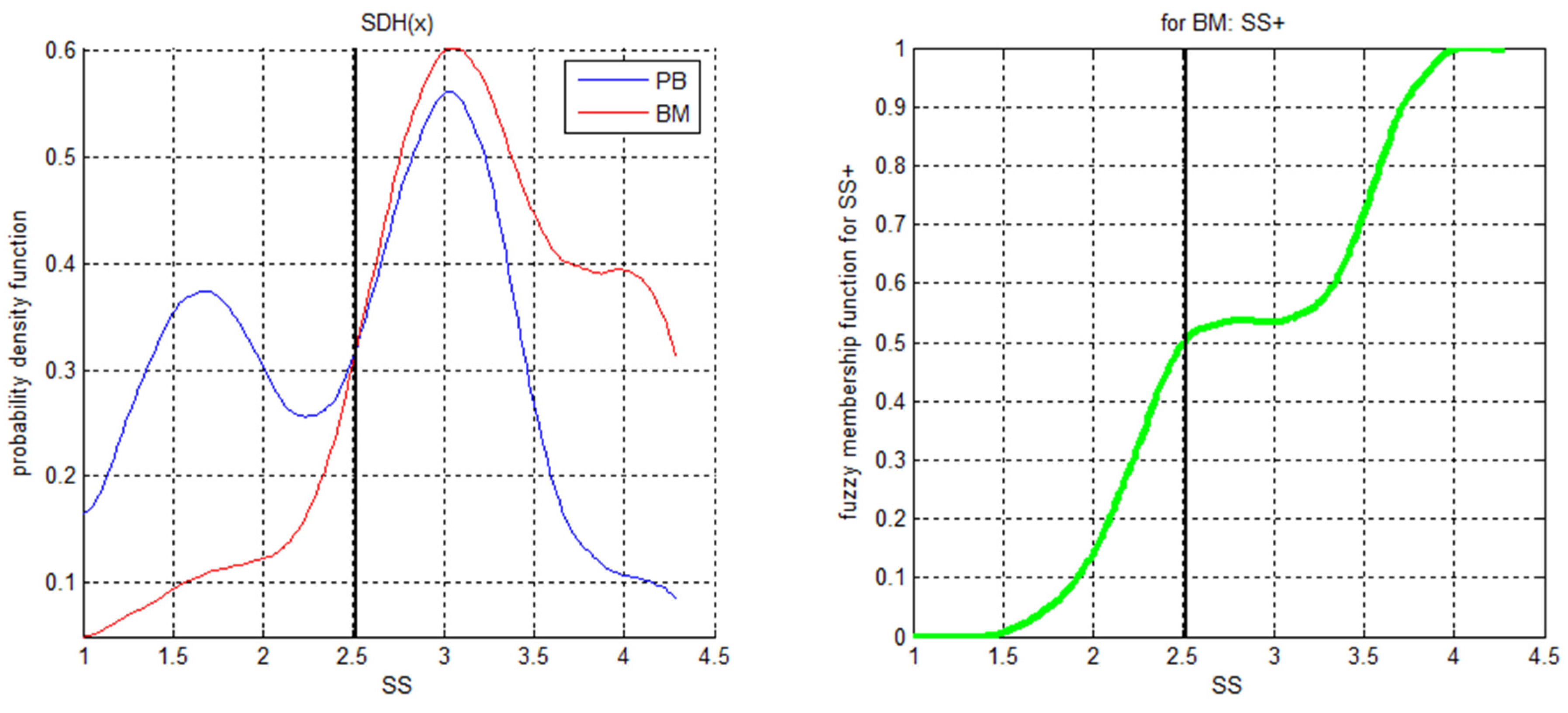
Table 5). For example, in ALPOD’s descriptions for the identified subpopulations “Progenitor B cells” and “Thrombocyte aggregations”, one of the terms in the description is “
“ (see
Table 4). The calculatable form of this expression is derived from the distribution as depicted in
Figure 3. The lower part of the distribution is the negation of
Figure 3 right panel, i.e., the smaller the SS value is compared to the Bayes decision limit of
the more the condition
is fulfilled.
In sum, blood stem cells were located in the BM niche and gave rise to myeloid progenitor cells (population 1). At the developmental maturation stage of granulocytes, these cells were released into the PB (populations 2 and 4). T cells were derived from the thymus and proliferated in the PB upon stimulation following infection (population 3). Therefore, these cell types predominantly occurred in the PB. Hematogenous cells are B-cell precursor cells that were found almost exclusively in the BM (population 5). However, due to active cell trafficking, there was no strict border between the two organic distributional spaces.
We further sought to verify the principle of the algorithm with another dataset and compared BM samples diagnosed as normal (healthy) BM (
N = 25) and samples with leukemia cell infiltration (
N = 25); see [
68] for a detailed description. The results are summarized in
Table 6 and visualized in
Figure 4.
Domain experts assess flow cytometric samples based on biological cell populations with characteristic fluorescence expressions in two-dimensional dot plots, as demonstrated for distinguishing leukemias from normal controls in
Figure 5. The results from data scientists can be presented in these two-dimensional dot plots, enabling their plausibility to be verified by domain experts. This approach integrates domain-specific knowledge into the analysis, ensuring that the interpretations are not only statistically valid but also biologically relevant and meaningful.
Within a computation time of several minutes, ALPODS identified a major dominant population labeled “blast cells” by the flow cytometry experts in the VisPanels of
Figure 6a,b. The population was described by one explanation, resulting in a
classification accuracy. The blast cell population was characterized by a high expression of the antigens CD34 and CD117, while CD45 was only moderately expressed, as depicted in a conventional scatter plot (
Figure 6b). “Blast cell” is another term for “leukemia cell”. A blast cell infiltration level
defines the disease “leukemia” as diagnosed out of a BM specimen by physicians. If a second population (magenta in the VisPanels in
Figure 6a,b) defined by an additional explanation is considered, the accuracy of the test data is
. The comparative approach FAUST [
32] identified
212 relevant populations after a computation time of
3 h. The application of RFs to these populations resulted in an accuracy of
on the test data. The results are summarized in
Table 6.
The Applications of ALPODS
The International Prognostic Index (IPI) is used to predict outcomes in chronic lymphocytic leukemia (CLL) based on five factors, including genetic analysis [
80]. We aimed to determine if multiparameter flow cytometry data could predict CLL outcomes using ALPODS and explain the results through distinctive cell populations. A total of 157 CLL cases were analyzed, and the ALPODS XAI algorithm was used to identify cell populations linked to poor outcomes (death or first-line treatment failure). ALPODS identified highly predictive cell populations using MPFC data, improving CLL outcome predictions [
81].
Minimal residual disease (MRD) detection is crucial for predicting survival and relapse in acute myeloid leukemia (AML) [
82]. However, the bone marrow (BM) dilution with peripheral blood (PB) increases with aspiration volume, causing underestimation of residual AML blast amount and potentially leading to false-negative MRD results [
83,
84]. To address this issue, we developed an automated method for the simultaneous measurement of hemodilution in BM samples and MRD levels [
85]. First, ALPODS was trained and validated using flow cytometric data from 126 BM and 23 PB samples from 35 patients. ALPODS identified discriminating populations, both specific and non-specific for BM or PB, and considered their frequency differences. Based on the identified populations, the subsequent Cinderella method accurately predicted PB dilution, showing a strong correlation with the Holdrinet formula [
86], and, hence, the methodology can help to prevent false-negative MRD reports.
6. Discussion
AI systems constructed by ML have shown in recent years that these algorithms are able to perform classification tasks with precision similar to that of experts in a domain. Subsymbolic systems, for example, ANNs, are usually the best-performing systems. However, the subsymbolic approach deliberately sacrifices explainability, i.e., human understanding, in a tradeoff with performance. For life-critical applications, such as medical diagnosis, AI systems that are able to explain their decisions (XAI systems) are needed. Here, XAI systems, which identify subpopulations within a dataset that are (first) relevant for the diagnosis process and (second) describable in the form of a set of conditions (rule) that are in principle human-understandable and genuinely reflect the clinical condition, are considered. For such systems, several criteria are relevant. First, from a practical standpoint, the algorithm should be able to finish its task in a reasonable amount of time on typical hardware. Second, the performance achieved in terms of decision accuracy should exceed 80–90%, and third and most importantly, the results must be understandable in the sense that they are interpretable by and explainable to human domain experts.
In this work, we introduced an XAI algorithm (ALPODS) designed for large (
) and multivariate data (d ≥ 10), such as typical flow cytometry data. The distinction between BM and PB served as an example because many approaches have been proposed to solve the problem of BM dilution with PB [
86,
87]. Holdrinet et al. [
86] suggested counting nucleated cells with an external hemacytometer device, while others have suggested running a separate flow cytometric panel to quantify the populations that predominantly occur in the BM or PB areas [
87,
88]. Of note, the ALPODS algorithm is capable of extracting information about population differences from both datasets, thus making additional external or additional measurements dispensable. In comparison with other state-of-the-art XAI algorithms developed for the same task, ALPODS delivered human-understandable results. Experts in flow cytometry identified the cell populations as “true” progenitor cells, T cells, or granulocytes. Moreover, experts verified that the distribution patterns of these populations in PB and BM were plausible according to the knowledge of hematologists. To achieve enhanced understandability, a visualization tool relying on differential density estimation between the cases in a subpopulation (partition) vs. the rest of the data was essential because it seems that in several domains, visualizations significantly improve the understandability of explanations. For example, in xDNN, appropriate training images are algorithmically selected as prototypes and combined into logical rules (called mega clouds) in the last layer to explain the CNN’s decision [
15]. Similar to the VisPanel of the ALPODS method introduced here, the presented rules are human-understandable because they are presented as images. However, the generalization of the explanation process is performed by neural networks’ well-known application of a Voronoi tessellation [
89,
90] of the projection produced by principal component analysis (PCA) [
91]. If a high-dimensional dataset, for example, N > 18,000 gene expressions of patients diagnosed with a variety of cancer illnesses, is considered [
72], neither the logical rules consisting of prototypes nor the Voronoi tessellation of PCA would be understandable to a domain expert. Moreover, it is well known that PCA does not reproduce high-dimensional structures appropriately [
89], which could lead to untrustworthy explanations.
The XAI expert system constructed using the results of ALPODS was able to correctly distinguish between two material sources by identifying cellular and subcellular events, i.e., BM and PB. In particular, every diagnosis provided by this XAI system could be understood and validated by human experts. Moreover, in line with the argument presented by Rudin [
9], the results indicated that a trade-off between understandability and accuracy is not necessary for XAI systems that use biomedical data.
Differentiating between PB and BM is manageable for human experts in the flow cytometry field without AI. Therefore, this problem is perfectly suited as a proof of concept for XAI because it is comprehensible and verifiable to human experts, as we showed here. However, the degree of BM dilution with PB increases with the aspiration volume, causing the residual AML blast amount to be consecutively underestimated. To prevent false-negative MRD results, ALPODS was already applied using a simple automated method for the one-tube simultaneous measurement of hemodilution in BM samples and MRD levels [
85].


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}