

Uncovering Disease-Related Polymorphisms through Correlations between SNP Frequencies, Population and Epidemiological Data

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
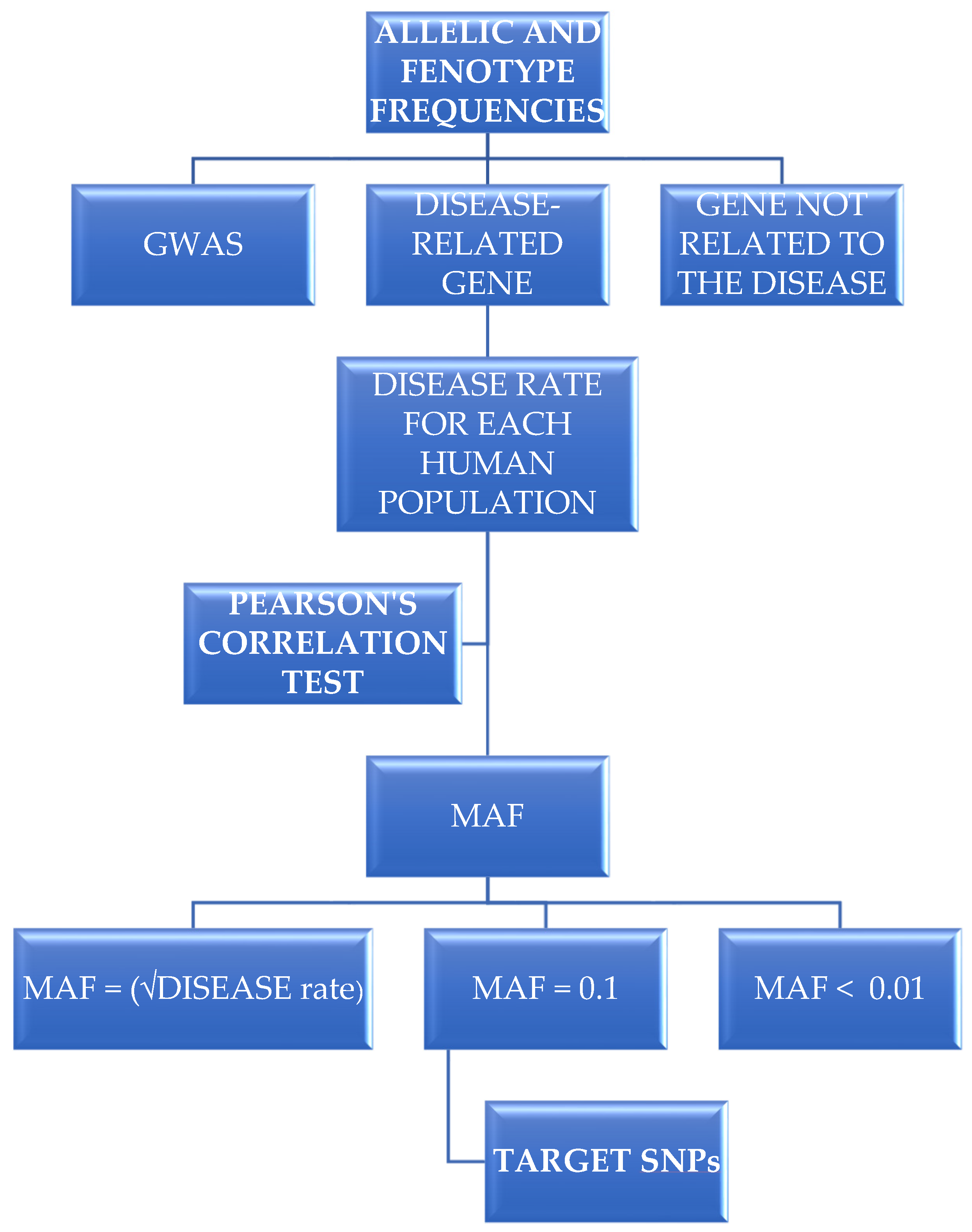
2.1. Choosing Allelic Variants and Obtaining Their Frequencies for Different Human Populations
2.2. Statistical Analysis
2.3. Linkage Disequilibrium
3. Results
3.1. The Correlation Approach Detected Significant Variants in GWAS-Associated Data But Not in the Postulated Unrelated Genes
3.2. Proof of Concept: The TPH2, NR3C1 and SLC6A3, but Not the SLC6A2 Gene Variants, Were Associated with Depression
3.3. Linkage Disequilibrium and Variant Effects on Traits and Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Barreiro, L.B.; Laval, G.; Quach, H.; Patin, E.; Quintana-Murci, L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008, 40, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; Alfirevic, A.; Pirmohamed, M. Pharmacogenomics: Current state-of-the-art. Genes 2014, 5, 430–443. [Google Scholar] [CrossRef] [Green Version]
- 1000 Genomes Project Consortium; Abecasis, G.R.C.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Xiao, H.; Yang, Y.; Cao, D.; Wang, L.; Yang, X.; Qiu, X.; Qiao, Z.; Song, J.; Liu, Y.; et al. Interaction of tryptophan hydroxylase 2 gene and life events in susceptibility to major depression in a Chinese Han population. J. Affect. Disord. 2015, 188, 304–309. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S. The HapMap and Genome-Wide Association Studies in Diagnosis and Therapy. Annu. Rev. Med. 2009, 60, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef]
- Wang, W.Y.S.; Barratt, B.J.; Clayton, D.G.; Todd, J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005, 6, 109–118. [Google Scholar] [CrossRef]
- Wellcome, T.; Case, T.; Consortium, C. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Bello, J.; Jiménez-Morales, M. Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases. Gac. Med. Mex. 2017, 153, 238–250. [Google Scholar]
- Ormel, J.; Hartman, C.A.; Snieder, H. The genetics of depression: Successful genome-wide association studies introduce new challenges. Transl. Psychiatry 2019, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amare, A.T.; Vaez, A.; Hsu, Y.-H.; Direk, N.; Kamali, Z.; Howard, D.M.; McIntosh, A.M.; Tiemeier, H.; Bültmann, U.; Snieder, H.; et al. Bivariate genome-wide association analyses of the broad depression phenotype combined with major depressive disorder, bipolar disorder or schizophrenia reveal eight novel genetic loci for depression. Mol. Psychiatry 2020, 25, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obenchain, V.; Lawrence, M.; Carey, V.; Gogarten, S.; Shannon, P.; Morgan, M. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 2014, 30, 2076–2078. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017; pp. 1–24. Available online: https://apps.who.int/iris/handle/10665/25461017 (accessed on 20 August 2021).
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [Green Version]
- Machiela, M.J.; Chanock, S.J. LDassoc: An online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 2018, 34, 887–889. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.; Zheng-Bradley, X.; Smith, R.; Kulesha, E.; Xiao, C.; Toneva, I.; Vaughan, B.; Preuss, D.; Leinonen, R.; Shumway, M.; et al. The 1000 Genomes Project: Data management and community access. Nat. Methods 2012, 9, 459–462. [Google Scholar] [CrossRef] [Green Version]
- Cardon, L.R.; Palmer, L.J. Population stratification and spurious allelic association. Lancet 2003, 361, 598–604. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaid, D.J.; Sinnwell, J.P.; Jenkins, G.D.; McDonnell, S.; Ingle, J.N.; Kubo, M.; Goss, P.E.; Costantino, J.P.; Wickerham, D.L.; Weinshilboum, R.M. Using the gene ontology to scan multilevel gene sets for associations in genome wide association studies. Genet. Epidemiol. 2012, 36, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Utge, S.; Soronen, P.; Partonen, T.; Loukola, A.; Kronholm, E.; Pirkola, S.; Nyman, E.; Porkka-Heiskanen, T.; Paunio, T. A population-based association study of candidate genes for depression and sleep disturbance. Am. J. Med. Genet. 2010, 153, 468–476. [Google Scholar] [CrossRef]
- de Araújo Pereira, P.; Romano-Silva, M.A.; Bicalho, M.A.C.; De Marco, L.; Correa, H.; de Campos, S.B.; de Moraes, E.N.; Torres, K.C.D.L.; de Souza, B.R.; de Miranda, D.M. Association Between Tryptophan Hydroxylase-2 Gene and Late-Onset Depression. Am. J. Geriatr. Psychiatry 2011, 19, 825–829. [Google Scholar] [CrossRef]
- Campos, S.B.; Miranda, D.M.; Souza, B.R.; Pereira, P.A.; Neves, F.S.; Tramontina, J.; Kapczinski, F.; Romano-Silva, M.A.; Correa, H. Association study of tryptophan hydroxylase 2 gene polymorphisms in bipolar disorder patients with panic disorder comorbidity. Psychiatr. Genet. 2011, 21, 106–111. [Google Scholar] [CrossRef]
- Gao, J.; Jia, M.; Qiao, D.; Qiu, H.; Sokolove, J.; Zhang, J.; Pan, Z. TPH2 gene polymorphisms and bipolar disorder: A meta-analysis. Am. J. Med. Genet. 2016, 171, 145–152. [Google Scholar] [CrossRef]
- Oades, R.D.; Lasky-Su, J.; Christiansen, H.; Faraone, S.V.; Sonuga-Barke, E.J.; Banaschewski, T.; Chen, W.; Anney, R.J.; Buitelaar, J.K.; Ebstein, R.P.; et al. The influence of serotonin- and other genes on impulsive behavioral aggression and cognitive impulsivity in children with attention-deficit hyperactivity disorder (ADHD): Findings from a family-based association test (FBAT) analysis. Behav. Brain Funct. 2008, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Chen, W.; Buitelaar, J.; Banaschewski, T.; Oades, R.D.; Franke, B.; Sonuga-Barke, E.; Ebstein, R.; Eisenberg, J.; Gill, M.; et al. Genetic heterogeneity in ADHD: DAT1 gene only affects probands without CD Genetic heterogeneity in ADHD: DAT1 gene only affects probands without CD. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 1481–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hack, L.M.; Kalsi, G.; Aliev, F.; Kuo, P.-H.; Prescott, C.A.; Patterson, D.G.; Walsh, D.; Dick, D.M.; Riley, B.P.; Kendler, K.S. Limited associations of dopamine system genes with alcohol dependence and related traits in the Irish Affected Sib Pair Study of Alcohol Dependence (IASPSAD). Alcohol. Clin. Exp. Res. 2011, 35, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Martelle, S.E.; Raffield, L.M.; Palmer, N.D.; Cox, A.J.; Freedman, B.I.; Hugenschmidt, C.E.; Williamson, J.D.; Bowden, D.W. Dopamine pathway gene variants may modulate cognitive performance in the DHS—Mind Study. Brain Behav. 2016, 6, e00446. [Google Scholar] [CrossRef]
- Tan, E.-C.; Chua, T.-E.; Lee, T.M.Y.; Tan, H.-S.; Ting, J.L.Y.; Chen, H.Y. Case-control study of glucocorticoid receptor and corticotrophin-releasing hormone receptor gene variants and risk of perinatal depression. BMC Pregnancy Childbirth 2015, 15, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, E.; Kang, J.; Kim, A.; Choi, S.; Han, K.-M.; Tae, W.S.; Chang, H.S.; Son, K.R.; Greenberg, T.; Joe, S.-H.; et al. Influence of BclI C/G (rs41423247) on hippocampal shape and white matter integrity of the parahippocampal cingulum in major depressive disorder. Psychoneuroendocrinology 2016, 72, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Zobel, A.; Jessen, F.; von Widdern, O.; Schuhmacher, A.; Höfels, S.; Metten, M.; Rietschel, M.; Scheef, L.; Block, W.; Becker, T.; et al. Unipolar depression and hippocampal volume: Impact of DNA sequence variants of the glucocorticoid receptor gene. Am. J. Med. Genet. 2008, 147, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Couto-Silva, C.M.; Nunes, K.; Venturini, G.; Araújo Castro e Silva, M.; Pereira, L.V.; Comas, D.; Pereira, A.; Hünemeier, T. Indigenous people from Amazon show genetic signatures of pathogen-driven selection. Sci. Adv. 2023, 9, eabo0234. [Google Scholar] [CrossRef]


{kind=link}
| Alternative Allele Frequency | Total | sigRR | sigAA | SNP (Allele Frequency/Correlation p-Value) * |
|---|---|---|---|---|
| <0.01 | 0 | 0 | 0 | ------- |
| 0.01–0.1 | 4 | 0 | 1 | rs17727765 (AFr = 0.03, p = 0.004); |
| 0.1–0.33 | 22 | 0 | 4 | rs7200826 (AFr = 0.15, p = 0.003); rs112348907 (AFr = 0.27, p = 0.007); rs2422320(AFr = 0.29, p = 0.0002); rs2422321 (AFr = 0.29, p = 0.0001) |
| 0.33–0.66 | 38 | 0 | 0 | ------- |
| 0.66–0.9 | 18 | 4 | 0 | rs10929355 (AFr = 0.69, p = 0.010); rs4904738 (AFr = 0.71, p = 0.002); rs7044150 (AFr = 0.84, p = 0.003); rs1354115 (AFr = 0.81, p = 0.004) |
| 0.9–0.99 | 0 | 0 | 0 | ------- |
| >0.99 | 0 | 0 | 0 | ------- |
| Alternative Allele Frequency | Total | sigRR | sigAA | SNP (Allele Frequency/Correlation p-Value) * |
|---|---|---|---|---|
| <0.01 | 103 | 0 | 0 | ------- |
| 0.01–0.1 | 162 | 0 | 0 | ------- |
| 0.1–0.33 | 101 | 0 | 1 | rs4760820 (AFr = 0.179. p = 0.002); |
| 0.33–0.66 | 122 | 0 | 5 | ------- |
| 0.66–0.9 | 57 | 0 | 0 | ------- |
| 0.9–0.99 | 3 | 1 | 0 | rs7298203 (AFr = 0.974. p = 0.008) |
| >0.99 | 0 | 0 | 0 | ------- |
| Alternative Allele Frequency | Total | sigRR | sigAA | SNP (Allele Frequency/Correlation p-Value) * |
|---|---|---|---|---|
| <0.01 | 225 | 2 | 3 | ------- |
| 0.01–0.1 | 167 | 0 | 11 | rs116798177 (AFr = 0.02, p = 0.004); rs142327762 (AFr = 0.04, p = 0.002); rrs61752263 (AFr = 0.04, p = 0.002); rs55817235 (AFr = 0.04, p = 0.002); rrs56150733 (AFr = 0.04, p = 0.002); rs72801051 (AFr = 0.05, p = 0.002); rrs72801054 (AFr = 0.05, p = 0.002); rs141755899 (AFr = 0.05, p = 0.002); rrs72801080 (AFr = 0.05, p = 0.002); rs10515522 (AFr = 0.05, p = 0.002); rrs72802806 (AFr = 0.09, p = 0.010) |
| 0.1–0.33 | 126 | 0 | 14 | rs258814 (AFr = 0.21. p = 0.004); rs13155635 (AFr = 0.28. p = 0.010); rs860457 (AFr = 0.21. p = 0.006); rs852979 (AFr = 0.21. p = 0.007); rs852982 (AFr = 0.21. p = 0.004); rs190488 (AFr = 0.21. p = 0.007); rs33380 (AFr = 0.21. p = 0.01); rs34158792 (AFr = 0.23. p = 0.020); rs61752282 (AFr = 0.21. p = 0.01); rs111440401 (AFr = 0.15. p = 0.008); rs1866388 (AFr = 0.21. p = 0.009); rs10053679 (AFr = 0.22. p = 0.012); rs41423247 (AFr = 0.25. p = 0.001); rs11747997 (AFr = 0.21. p = 0.009); |
| 0.33–0.66 | 27 | 0 | 0 | ------- |
| 0.66–0.9 | 6 | 1 | 0 | rs1837262 (AFr = 0.77. p = 0.01) |
| 0.9–0.99 | 11 | 0 | 0 | ------- |
| >0.99 | 0 | 0 | 0 | ------- |
| Alternative Allele Frequency | Total | sigRR | sigAA | SNP (Allele Frequency/Correlation p-Value) * |
|---|---|---|---|---|
| <0.01 | 40 | 0 | 0 | ------- |
| 0.01–0.1 | 183 | 0 | 0 | ------- |
| 0.1–0.33 | 80 | 0 | 4 | rs13189021 (AFr = 0.08, p = 0.001); rs10052016 (AFr = 0.18, p = 0.002); rs62331084 (AFr = 0.09, p = 0.003); rs10053602 (AFr = 0.18, p = 0.007); |
| 0.33–0.66 | 66 | 0 | 0 | ------- |
| 0.66–0.9 | 5 | 0 | 0 | ------- |
| 0.9–0.99 | 2 | 0 | 0 | ------- |
| >0.99 | 1 | 0 | 0 | ------- |
| Gene | Linkage SNPs | |
|---|---|---|
| TPH2 | ----- | |
| NR3C1 | Block 1 | rs10053679, rs34158792, rs860457, rs852979, rs258814, rs852982, rs190488, rs33380, rs61752282, rs1866388, rs11747997, rs1837262 |
| Block 2 | rs142327762, rs61752263, rs55817235, rs56150733, rs72801051, rs72801054, rs141755899, rs72801080, rs10515522 | |
| SLC6A3 | rs10052016; rs10053602 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis, S.M.D.; Bugs, C.A.; Chies, J.A.B.; Cañedo, A.D. Uncovering Disease-Related Polymorphisms through Correlations between SNP Frequencies, Population and Epidemiological Data. BioMedInformatics 2023, 3, 467-477. https://doi.org/10.3390/biomedinformatics3020032
Reis SMD, Bugs CA, Chies JAB, Cañedo AD. Uncovering Disease-Related Polymorphisms through Correlations between SNP Frequencies, Population and Epidemiological Data. BioMedInformatics. 2023; 3(2):467-477. https://doi.org/10.3390/biomedinformatics3020032
Chicago/Turabian StyleReis, Samara Marques Dos, Cristhian Augusto Bugs, José Artur Bogo Chies, and Andrés Delgado Cañedo. 2023. "Uncovering Disease-Related Polymorphisms through Correlations between SNP Frequencies, Population and Epidemiological Data" BioMedInformatics 3, no. 2: 467-477. https://doi.org/10.3390/biomedinformatics3020032