
Trecode: A FAIR Eco-System for the Analysis and Archiving of Omics Data in a Combined Diagnostic and Research Setting

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Softwares
2.2. Datamodel
2.3. Identifiers
2.4. Workflow Language
3. Results
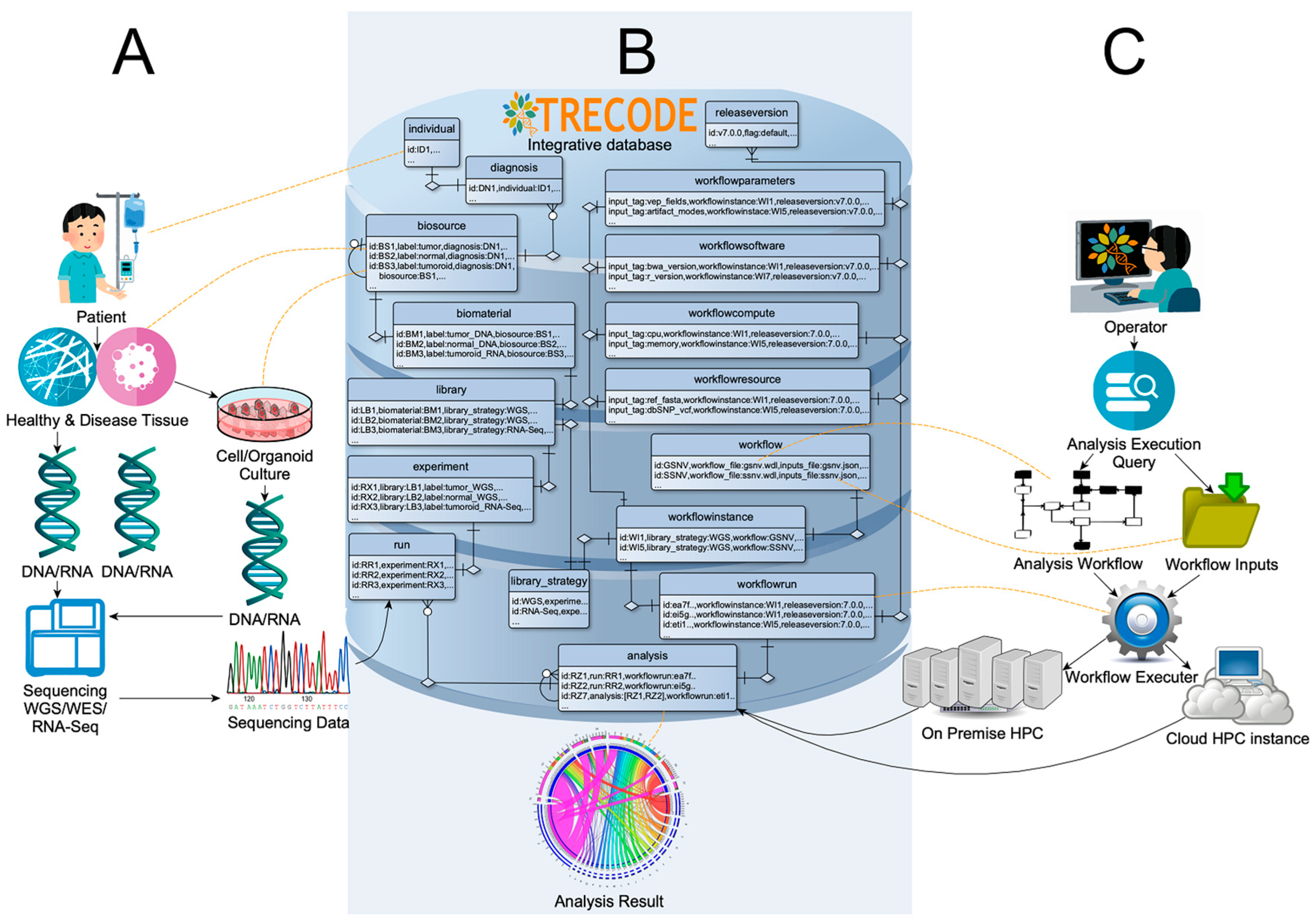
3.1. Platform Design
3.2. Data Model
3.3. Accessibility to Users from Multiple Disciplines
3.3.1. Operators
3.3.2. Wet-Lab Scientists
3.3.3. Bioinformaticians
3.3.4. Data Security
3.4. Data Governance
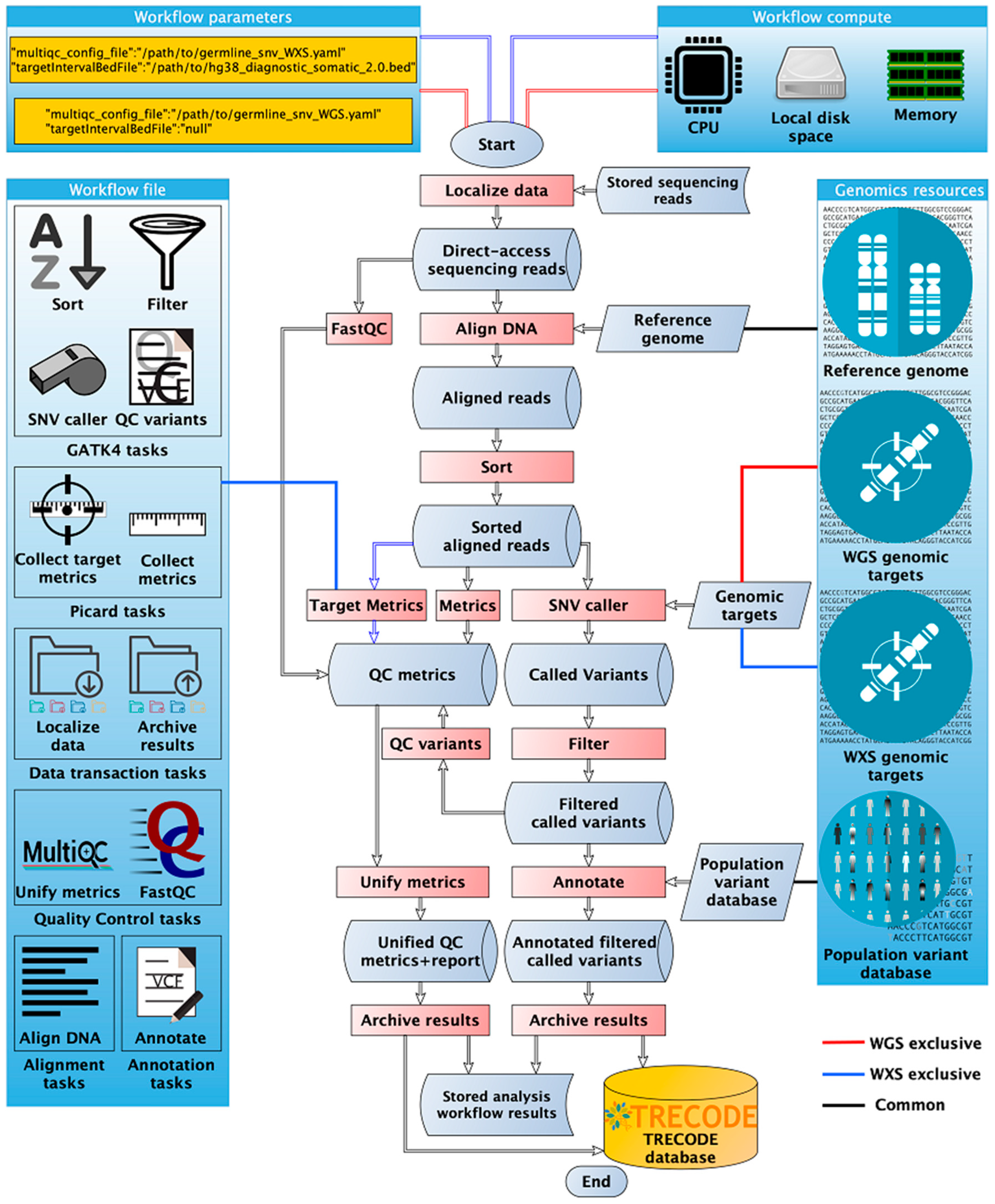
3.5. Data Analysis
3.5.1. Integrated Data Analysis Workflows
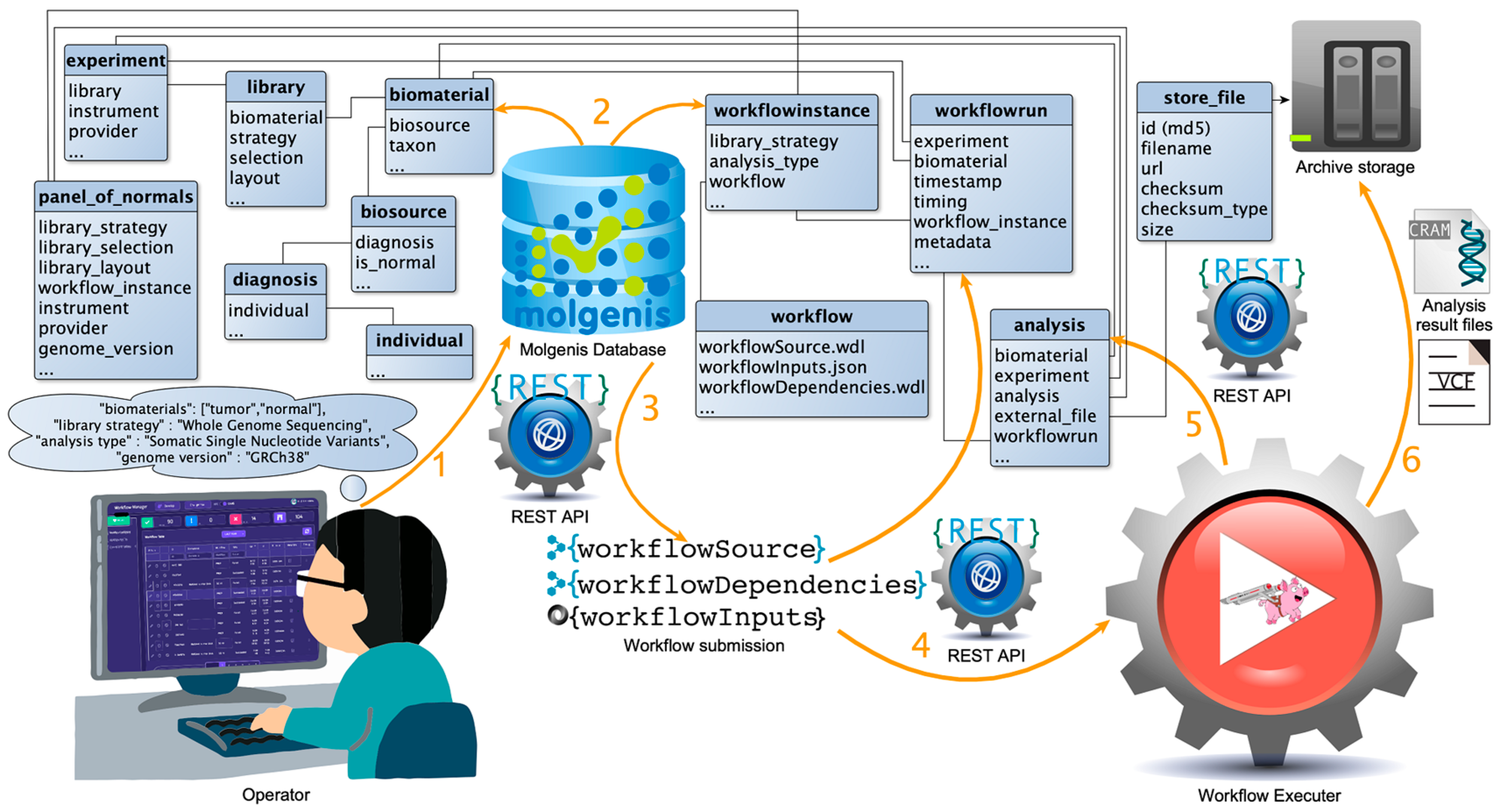
3.5.2. Analyses Reproducibility by Automation and Tracking System
3.5.3. Workflow Manager
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Höllein, A.; Twardziok, S.O.; Walter, W.; Hutter, S.; Baer, C.; Hernandez-Sanchez, J.M.; Meggendorfer, M.; Haferlach, T.; Kern, W.; Haferlach, C. The Combination of WGS and RNA-Seq Is Superior to Conventional Diagnostic Tests in Multiple Myeloma: Ready for Prime Time? Cancer Genet. 2020, 242, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smadbeck, J.; Peterson, J.F.; Pearce, K.E.; Pitel, B.A.; Figueroa, A.L.; Timm, M.; Jevremovic, D.; Shi, M.; Stewart, A.K.; Braggio, E.; et al. Mate Pair Sequencing Outperforms Fluorescence in Situ Hybridization in the Genomic Characterization of Multiple Myeloma. Blood Cancer J. 2019, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrochristos, I.D.; Ziogas, D.E.; Goussia, A.; Glantzounis, G.K.; Roukos, D.H. Bulk and Single-Cell Next-Generation Sequencing: Individualizing Treatment for Colorectal Cancer. Cancers 2019, 11, 1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Sinha, C.; Chinnaiyan, A.M. Precision Oncology in the Age of Integrative Genomics. Nat. Biotechnol. 2018, 36, 46–60. [Google Scholar] [CrossRef]
- Seibel, N.L.; Janeway, K.; Allen, C.E.; Chi, S.N.; Cho, Y.-J.; Glade Bender, J.L.; Kim, A.; Laetsch, T.W.; Irwin, M.S.; Takebe, N.; et al. Pediatric Oncology Enters an Era of Precision Medicine. Curr. Probl. Cancer 2017, 41, 194–200. [Google Scholar] [CrossRef]
- Kulkarni, P.; Frommolt, P. Challenges in the Setup of Large-Scale Next-Generation Sequencing Analysis Workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef]
- Frazer, S. Workflow Description Language. 2014. Available online: https://software.broadinstitute.org/wdl/ (accessed on 14 September 2020).
- Amstutz, P. Common Workflow Language. 2016. Available online: https://github.com/common-workflow-language/common-workflow-language (accessed on 14 September 2020).
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil Enables Reproducible, Open Source, Big Biomedical Data Analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Cromwell Homepage. Available online: https://cromwell.readthedocs.io/en/stable/ (accessed on 14 September 2020).
- Fjukstad, B.; Bongo, L.A. A Review of Scalable Bioinformatics Pipelines. Data Sci. Eng. 2017, 2, 245–251. [Google Scholar] [CrossRef] [Green Version]
- van der Velde, K.J.; Imhann, F.; Charbon, B.; Pang, C.; van Enckevort, D.; Slofstra, M.; Barbieri, R.; Alberts, R.; Hendriksen, D.; Kelpin, F.; et al. MOLGENIS Research: Advanced Bioinformatics Data Software for Non-Bioinformaticians. Bioinformatics 2019, 35, 1076–1078. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tarraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European Nucleotide Archive. Nucleic Acids Res. 2011, 39, D28–D31. [Google Scholar] [CrossRef]
- Fielding, R.T. Architectural Styles and the Design of Network-Based Software Architectures; University of California: Irvine, CA, USA, 2000. [Google Scholar]
- Shumway, M.; Cochrane, G.; Sugawara, H. Archiving next Generation Sequencing Data. Nucleic Acids Res. 2010, 38, D870–D871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Beltrán, A.; Maguire, E.; Sansone, S.-A.; Rocca-Serra, P. LinkedISA: Semantic Representation of ISA-Tab Experimental Metadata. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, E.L.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; et al. Principles of Metadata Organization at the ENCODE Data Coordination Center. Database 2016, 2016, baw001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homepage Ga4gh/Large-Scale-Genomics-Wiki. Available online: https://github.com/ga4gh/large-scale-genomics-wiki (accessed on 14 December 2022).
- NCBO BioPortal. Available online: https://bioportal.bioontology.org/visits (accessed on 14 December 2022).
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Imran, M.; Hlavacs, H.; Haq, I.U.; Jan, B.; Khan, F.A.; Ahmad, A. Provenance Based Data Integrity Checking and Verification in Cloud Environments. PLoS ONE 2017, 12, e0177576. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Global Alliance for Genomics & Health SAM/BAM and Related Specifications. 2014. Available online: http://samtools.github.io/hts-specs/ (accessed on 14 December 2022).
- Hierarchical Data Format version 5. Available online: https://portal.hdfgroup.org/display/HDF5/HDF5 (accessed on 28 August 2020).
- GATK Best Practices. Available online: https://software.broadinstitute.org/gatk/best-practices/about (accessed on 28 August 2020).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Merkel, D. Docker: Lightweight Linux Containers for Consistent Development and Deployment. Linux J. 2014, 239, 2. [Google Scholar]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- El-Sappagh, S.; Franda, F.; Ali, F.; Kwak, K.-S. SNOMED CT Standard Ontology Based on the Ontology for General Medical Science. BMC Med. Inf. Decis Mak 2018, 18, 76. [Google Scholar] [CrossRef] [Green Version]
- Golbeck, J.; Fragoso, G.; Hartel, F.; Hendler, J.; Oberthaler, J.; Parsia, B. The National Cancer Institute’s Thesaurus and Ontology. SSRN Journal 2003. [Google Scholar] [CrossRef]
- Bandrowski, A.; Brinkman, R.; Brochhausen, M.; Brush, M.H.; Bug, B.; Chibucos, M.C.; Clancy, K.; Courtot, M.; Derom, D.; Dumontier, M.; et al. The Ontology for Biomedical Investigations. PLoS ONE 2016, 11, e0154556. [Google Scholar] [CrossRef] [Green Version]
- Malone, J.; Holloway, E.; Adamusiak, T.; Kapushesky, M.; Zheng, J.; Kolesnikov, N.; Zhukova, A.; Brazma, A.; Parkinson, H. Modeling Sample Variables with an Experimental Factor Ontology. Bioinformatics 2010, 26, 1112–1118. [Google Scholar] [CrossRef] [Green Version]
- Ison, J.; Kalas, M.; Jonassen, I.; Bolser, D.; Uludag, M.; McWilliam, H.; Malone, J.; Lopez, R.; Pettifer, S.; Rice, P. EDAM: An Ontology of Bioinformatics Operations, Types of Data and Identifiers, Topics and Formats. Bioinformatics 2013, 29, 1325–1332. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, V.; Ceol, A.; Ogier, A.G.E.; de Pretis, S.; Galeota, E.; Kishore, K.; Bora, P.; Croci, O.; Campaner, S.; Amati, B.; et al. Integrated Systems for NGS Data Management and Analysis: Open Issues and Available Solutions. Front. Genet. 2016, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Ko, G.; Kim, P.-G.; Yoon, J.; Han, G.; Park, S.-J.; Song, W.; Lee, B. Closha: Bioinformatics Workflow System for the Analysis of Massive Sequencing Data. BMC Bioinform. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Terra Cloud-Native Platform for Biomedical Researchers. Available online: https://app.terra.bio/ (accessed on 4 November 2020).
- Reisinger, E.; Genthner, L.; Kerssemakers, J.; Kensche, P.; Borufka, S.; Jugold, A.; Kling, A.; Prinz, M.; Scholz, I.; Zipprich, G.; et al. OTP: An Automatized System for Managing and Processing NGS Data. J. Biotechnol. 2017, 261, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wagle, P.; Nikolić, M.; Frommolt, P. QuickNGS Elevates Next-Generation Sequencing Data Analysis to a New Level of Automation. BMC Genom. 2015, 16, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R2 Bioinformatics Platform. Available online: http://R2.Amc.Nl (accessed on 7 September 2020).
- van der Velde, K.J.; Singh, G.; Kaliyaperumal, R.; Liao, X.; de Ridder, S.; Rebers, S.; Kerstens, H.H.D.; de Andrade, F.; van Reeuwijk, J.; De Gruyter, F.E.; et al. FAIR Genomes Metadata Schema Promoting Next Generation Sequencing Data Reuse in Dutch Healthcare and Research. Sci Data 2022, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- RedHat Ansible Is Simple IT Automation. Available online: https://www.ansible.com (accessed on 14 December 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role | Vocabularies Ontologies | Workflows Parameters | Samples Metadata | Genomics Analysis | System | Trecode Users |
|---|---|---|---|---|---|---|
| Admin | R + W | R + W | R + W | R + W | R + W | Admins |
| Manager | R + W | R + W | R + W | R + W | R | bioinformaticians |
| Editor | R | R | R + W | R + W | - | operators, bioinformaticians |
| Viewer | R | R | R | R | - | (wet-lab) researchers |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerstens, H.H.; Hehir-Kwa, J.Y.; van de Geer, E.; van Run, C.; Badloe, S.; Janse, A.; Baker-Hernandez, J.; de Vos, S.; van der Leest, D.; Verwiel, E.T.; et al. Trecode: A FAIR Eco-System for the Analysis and Archiving of Omics Data in a Combined Diagnostic and Research Setting. BioMedInformatics 2023, 3, 1-16. https://doi.org/10.3390/biomedinformatics3010001
Kerstens HH, Hehir-Kwa JY, van de Geer E, van Run C, Badloe S, Janse A, Baker-Hernandez J, de Vos S, van der Leest D, Verwiel ET, et al. Trecode: A FAIR Eco-System for the Analysis and Archiving of Omics Data in a Combined Diagnostic and Research Setting. BioMedInformatics. 2023; 3(1):1-16. https://doi.org/10.3390/biomedinformatics3010001
Chicago/Turabian StyleKerstens, Hindrik HD, Jayne Y Hehir-Kwa, Ellen van de Geer, Chris van Run, Shashi Badloe, Alex Janse, John Baker-Hernandez, Sam de Vos, Douwe van der Leest, Eugène TP Verwiel, and et al. 2023. "Trecode: A FAIR Eco-System for the Analysis and Archiving of Omics Data in a Combined Diagnostic and Research Setting" BioMedInformatics 3, no. 1: 1-16. https://doi.org/10.3390/biomedinformatics3010001