
UV-Induced Benzyloxy Rotamerization in an Ortho OH-Substituted Aryl Schiff Base





Abstract
:1. Introduction
2. Experimental and Computational Methods
2.1. Synthesis Procedure and Characterization
2.2. Matrix-Isolation and UV-Irradiation Experiments
2.3. Quantum Chemical Calculations
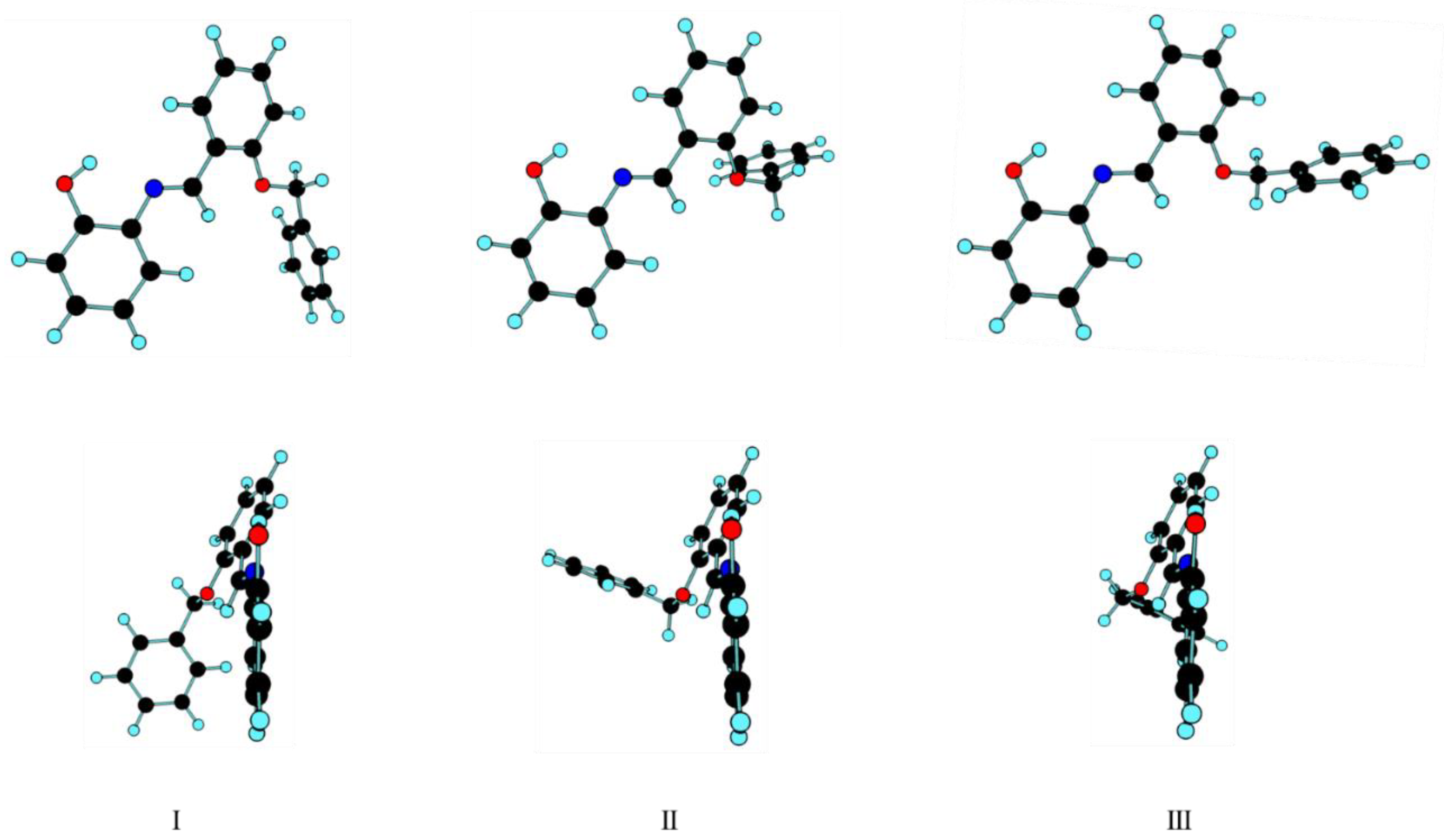
3. Results and Discussion
3.1. Geometrical Features and Energies of BBAP Isomeric Structures
3.2. Infrared Spectrum of Matrix-Isolated BBAP
3.3. Narrowband UV-Induced Rotamerization of Phenyl Ring
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borisova, N.E.; Reshetova, M.D.; Ustynyuk, Y.A. Metal-Free Methods in the Synthesis of Macrocyclic Schiff Bases. Chem. Rev. 2007, 107, 46–79. [Google Scholar] [CrossRef] [PubMed]
- Vigato, P.A.; Tamburini, S.; Bertolo, L. The development of compartmental macrocyclic Schiff bases and related polyamine derivatives. Coord. Chem. Rev. 2007, 251, 1311–1492. [Google Scholar] [CrossRef]
- Emregül, K.C.; Düzgün, E.; Atakol, O. The application of some polydentate Schiff base compounds containing aminic nitrogens as corrosion inhibitors for mild steel in acidic media. Corros. Sci. 2006, 48, 3243–3260. [Google Scholar] [CrossRef]
- Drozdzak, R.; Allaert, B.; Ledoux, N.; Dragutan, I.; Dragutan, V.; Verpoort, F. Ruthenium complexes bearing bidentate Schiff base ligands as efficient catalysts for organic and polymer syntheses. Coord. Chem. Rev. 2005, 249, 3055–3074. [Google Scholar] [CrossRef]
- Sessler, J.L.; Melfi, P.J.; Pantos, G.D. Uranium complexes of multidentate N-donor ligands. Coord. Chem. Rev. 2006, 250, 816–843. [Google Scholar] [CrossRef]
- Yang, C.-J.; Jenekhe, S.A. Conjugated aromatic polyimines. 2. Synthesis, structure, and properties of new aromatic polyazomethines. Macromolecules 1995, 28, 1180–1196. [Google Scholar] [CrossRef]
- Destri, S.; Khotina, I.A.; Porzio, W. 3-Hexyl Tetra-Substituted Sesquithienylene- Phenylene Polyazomethines with High Molecular Weight. Mechanistic Considerations. Macromolecules 1998, 31, 1079–1086. [Google Scholar] [CrossRef]
- Grigoras, M.; Catanescu, O.; Simionescu, C.I. Poly(azomethine)s. Rev. Roum. Chim. 2001, 46, 927–939. [Google Scholar]
- Kaya, I.; Vilayetoglu, A.R.; Mart, H. The synthesis and properties of oligosalicylaldehyde and its Schiff base oligomers. Polymer 2001, 42, 4859–4865. [Google Scholar] [CrossRef]
- Wu, P.; Bhamidipati, M.; Coles, M.; Rao, D. Biological nano-ceramic materials for holographic data storage. Chem. Phys. Lett. 2004, 400, 506–510. [Google Scholar] [CrossRef]
- Yuan, W.; Sun, L.; Tang, H.; Wen, Y.; Jiang, G.; Huang, W.; Jiang, L.; Song, Y.; Tian, H.; Zhu, D. A novel thermally stable spironaphthoxazine and its application in rewritable high density optical data storage. Adv. Mater. 2005, 17, 156–160. [Google Scholar] [CrossRef]
- Yanez, C.O.; Andrade, C.D.; Yao, S.; Luchita, G.; Bondar, M.V.; Belfield, K.D. Photosensitive polymeric materials for two-photon 3D WORM optical data storage systems. ACS Appl. Mater. Interfaces 2009, 1, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Melloni, A.; Rossi Paccani, R.; Donati, D.; Zanirato, V.; Sinicropi, A.; Parisi, M.L.; Martin, E.; Ryazantsev, M.; Ding, W.J.; Frutos, L.M. Modeling, preparation, and characterization of a dipole moment switch driven by Z/E photoisomerization. J. Am. Chem. Soc. 2010, 132, 9310–9319. [Google Scholar] [CrossRef]
- Staykov, A.; Watanabe, M.; Ishihara, T.; Yoshizawa, K. Photoswitching of conductance through salicylidene methylamine. J. Phys. Chem. C 2014, 118, 27539–27548. [Google Scholar] [CrossRef]
- Dalapati, S.; Jana, S.; Guchhait, N. Anion recognition by simple chromogenic and chromo-fluorogenic salicylidene Schiff base or reduced-Schiff base receptors. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 129, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, J.; Yoon, J. Recent advances in development of chiral fluorescent and colorimetric sensors. Chem. Rev. 2014, 114, 4918–4959. [Google Scholar] [CrossRef]
- Hadjoudis, E.; Mavridis, I.M. Photochromism and thermochromism of Schiff bases in the solid state: Structural aspects. Chem. Soc. Rev. 2004, 33, 579–588. [Google Scholar] [CrossRef]
- Amimoto, K.; Kawato, T. Photochromism of organic compounds in the crystal state. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 207–226. [Google Scholar] [CrossRef]
- Larkin, D.R. The role of catalysts in the air oxidation of aliphatic aldehydes. J. Org. Chem. 1990, 55, 1563–1568. [Google Scholar] [CrossRef]
- Vančo, J.; Švajlenová, O.; Račanská, E.; Muselík, J.; Valentová, J. Antiradical activity of different copper (II) Schiff base complexes and their effect on alloxan-induced diabetes. J. Trace Elem. Med. Biol. 2004, 18, 155–161. [Google Scholar] [CrossRef]
- Jarząbek, B.; Kaczmarczyk, B.; Sęk, D. Characteristic and spectroscopic properties of the Schiff-base model compounds. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2009, 74, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Raczuk, E.; Dmochowska, B.; Samaszko-Fiertek, J.; Madaj, J. Different Schiff Bases—Structure, Importance and Classification. Molecules 2022, 27, 787. [Google Scholar] [CrossRef] [PubMed]
- Król-Starzomska, I.; Filarowski, A.; Rospenk, M.; Koll, A.; Melikova, S. Proton Transfer Equilibria in Schiff Bases with Steric Repulsion. J. Phys. Chem. A 2004, 108, 2131–2138. [Google Scholar] [CrossRef]
- Minkin, V.I.; Tsukanov, A.V.; Dubonosov, A.D.; Bren, V.A. Tautomeric Schiff bases: Iono-, solvato-, thermo-and photochromism. J. Mol. Struct. 2011, 998, 179–191. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Pirbudak, G.; Berber, H.; Sıdır, İ. Study on the electronic and photophysical properties of the substitute-((2-phenoxybenzylidene) amino) phenol derivatives: Synthesis, solvatochromism, electric dipole moments and DFT calculations. J. Mol. Liq. 2017, 242, 1096–1110. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Berber, H.; Sıdır, İ. The Dipole Moments and Solvatochromism of ((4-(Benzyloxy) benzylidene) amino) phenol Compounds as Solvatochromic Materials. J. Solut. Chem. 2019, 48, 775–806. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Aslan, C.; Berber, H.; Sıdır, İ. The electronic structure, solvatochromism, and electric dipole moments of new Schiff base derivatives using absorbance and fluorescence spectra. Struct. Chem. 2019, 30, 835–851. [Google Scholar] [CrossRef]
- Avadanei, M.; Cozan, V.; Shova, S.; Paixão, J.A. Solid state photochromism and thermochromism of two related N-salicylidene anilines. Chem. Phys. 2014, 444, 43–51. [Google Scholar] [CrossRef]
- Sıdır, İ.; Sıdır, Y.G.; Góbi, S.; Berber, H.; Ildiz, G.O.; Fausto, R. UV-induced –OCH3 rotamerization in a matrix-isolated methoxy-substituted ortho-hydroxyaryl Schiff base. Photochem. Photobiol. Sci. 2022. [Google Scholar] [CrossRef]
- Sıdır, İ.; Gülseven Sıdır, Y.; Góbi, S.; Berber, H.; Fausto, R. Structural Relevance of Intramolecular H-Bonding in Ortho-Hydroxyaryl Schiff Bases: The Case of 3-(5-bromo-2-hydroxybenzylideneamino) Phenol. Molecules 2021, 26, 2814. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com/ (accessed on 3 January 2022).
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Reva, I.D.; Stepanian, S.G.; Adamowicz, L.; Fausto, F. Missing Conformers: Comparative Study of Conformational Cooling in Cyanoacetic Acid and Methyl Cyanoacetate Isolated in Low Temperature Inert Gas Matrixes. Chem. Phys. Lett. 2003, 374, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Rosado, M.T.S.; Lopes Jesus, A.J.; Reva, I.D.; Fausto, R.; Redinha, J.S. Conformational Cooling Dynamics in Matrix-Isolated 1,3-butanediol. J. Phys. Chem. A 2009, 113, 7499–7507. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformer | ΔEel | ΔE(0) | ΔG298.15 | P298.15 (%) |
|---|---|---|---|---|
| I | 0.00 | 0.00 | 0.00 | 89 |
| II | 3.52 | 3.91 | 6.83 | 6 |
| III | 3.65 | 4.16 | 7.13 | 5 |
| IV | 13.29 | 13.01 | 14.63 | |
| V | 15.23 | 14.58 | 14.09 | |
| VI | 12.83 | 12.23 | 14.09 | |
| VII | 14.37 | 14.14 | 16.80 | |
| VIII | 21.64 | 21.17 | 25.05 | |
| IX | 21.88 | 20.57 | 24.27 | |
| X | 22.09 | 20.76 | 22.50 | |
| XI | 28.74 | 27.18 | 27.92 |
| C–N=C–C | H–O–C–CN | CH–C–N=C | N=C–C–CH | CH–C–O–C | C–O–CH2–C | O–CH2–C–C | |
|---|---|---|---|---|---|---|---|
| Conformer | α | β | γ | δ | ε | ζ | |
| I | –177.4 | –2.7 | 21.7 | 6.7 | –0.7 | –179.5 | 80.1/–98.9 |
| II | –177.0 | –3.1 | 23.4 | 8.3 | 5.6 | –78.7 | –31.5/151.4 |
| III | –177.5 | –2.9 | 22.4 | 4.9 | –6.7 | 79.3 | 29.5/–153.3 |
| IV | –176.4 | –3.2 | 22.8 | 13.6 | –95.8 | 74.2 | 68.6/–111.2 |
| V | –179.1 | –2.3 | 16.4 | –3.0 | 87.0 | –77.8 | –64.6/115.5 |
| VI | –179.3 | –3.2 | 16.2 | 179.3 | –0.3 | –179.3 | –84.2/95.1 |
| VII | –179.2 | –1.5 | 12.4 | 177.1 | 2.4 | –77.6 | –30.8/152.1 |
| VIII | 179.9 | –1.7 | 25.9 | –175.3 | –105.4 | 74.2 | 68.7/–111.0 |
| IX | –179.4 | –3.9 | 28.1 | 179.5 | –98.5 | 174.9 | –27.3/154.7 |
| X | –179.7 | –3.2 | 29.7 | –177.1 | –100.1 | 170.5 | –67.4/112.8 |
| XI | –177.9 | –2.1 | 30.9 | –152.6 | 72.2 | –169.4 | 88.6/–90.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sıdır, İ.; Gülseven Sıdır, Y.; Góbi, S.; Berber, H.; Fausto, R. UV-Induced Benzyloxy Rotamerization in an Ortho OH-Substituted Aryl Schiff Base. Photochem 2022, 2, 376-389. https://doi.org/10.3390/photochem2020026
Sıdır İ, Gülseven Sıdır Y, Góbi S, Berber H, Fausto R. UV-Induced Benzyloxy Rotamerization in an Ortho OH-Substituted Aryl Schiff Base. Photochem. 2022; 2(2):376-389. https://doi.org/10.3390/photochem2020026
Chicago/Turabian StyleSıdır, İsa, Yadigar Gülseven Sıdır, Sándor Góbi, Halil Berber, and Rui Fausto. 2022. "UV-Induced Benzyloxy Rotamerization in an Ortho OH-Substituted Aryl Schiff Base" Photochem 2, no. 2: 376-389. https://doi.org/10.3390/photochem2020026