

Multifactorial Distress, the Warburg Effect, and Respiratory and pH Imbalance in Cancer Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress
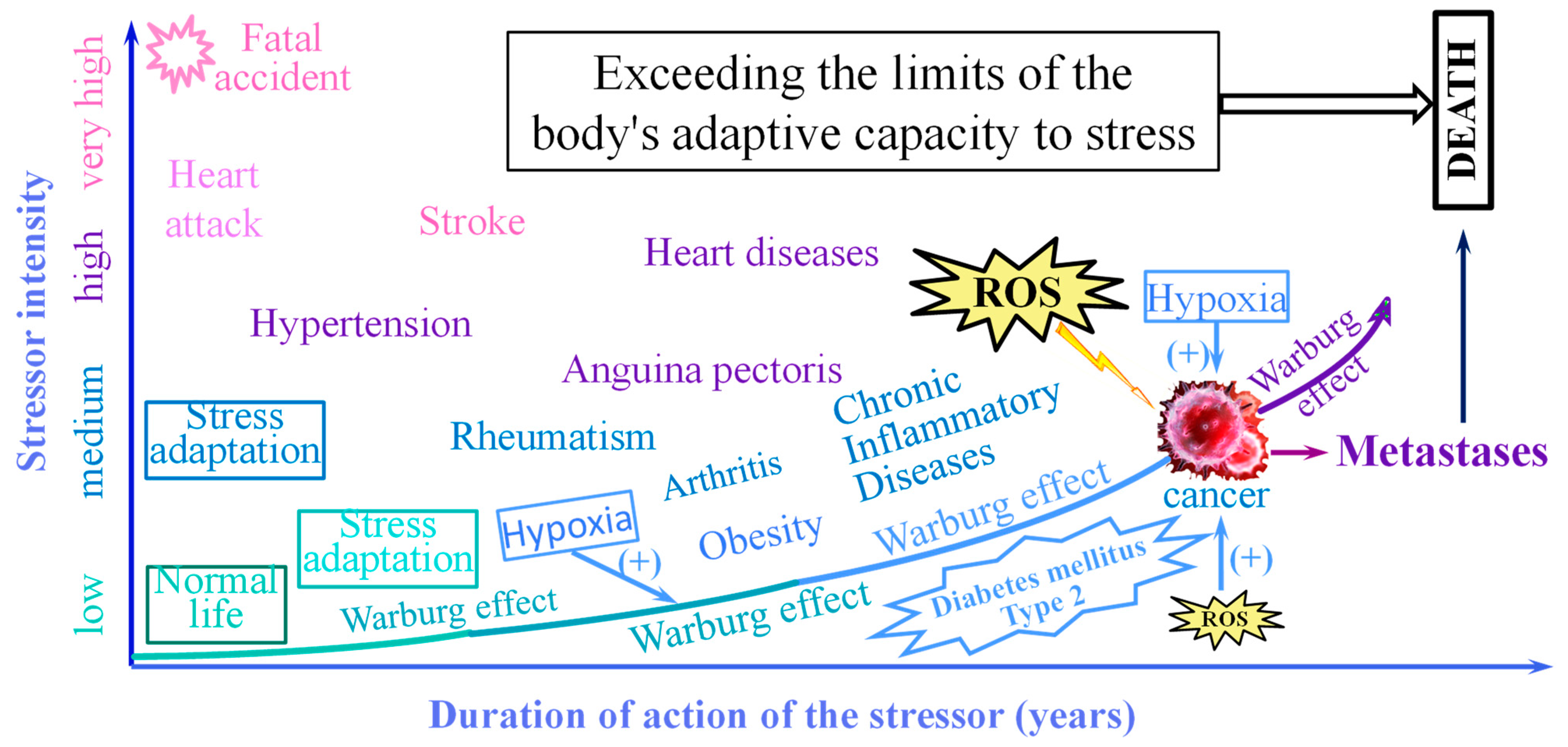
3. The Non-Specific Stress
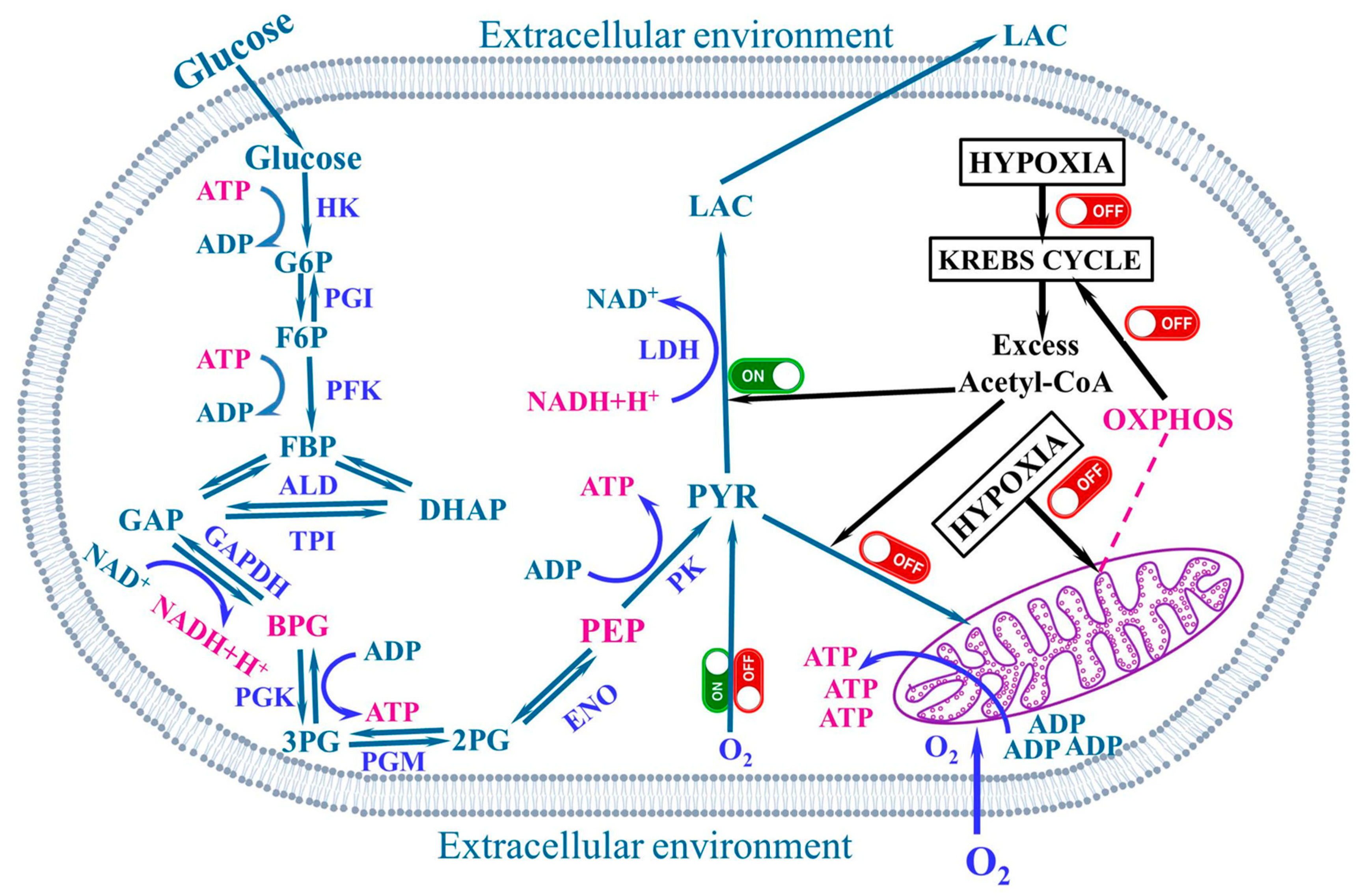
4. The Warburg Effect
5. The Aerobic Glycolysis
6. Copper and Cancer
7. Cancer and Lifestyle
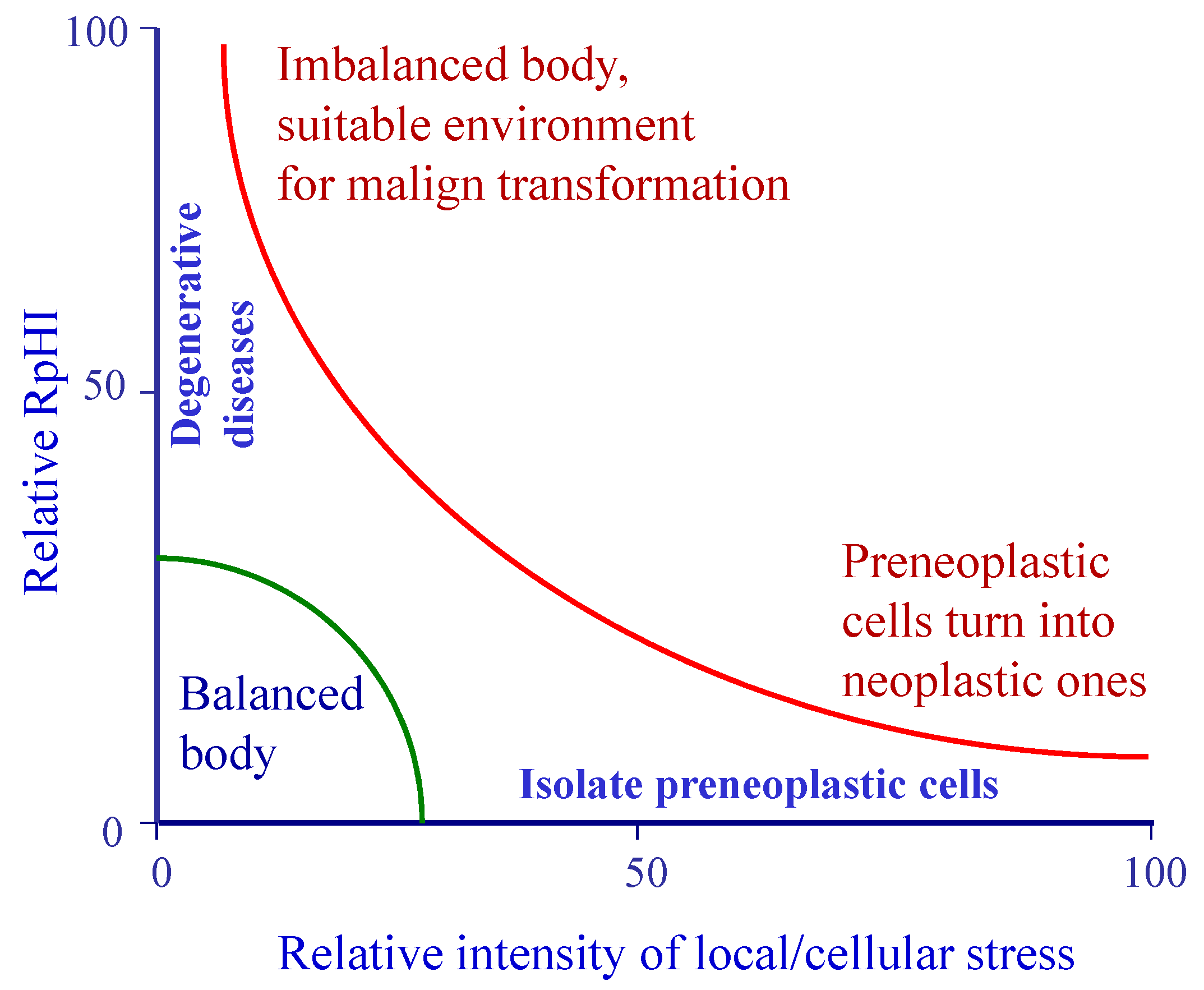
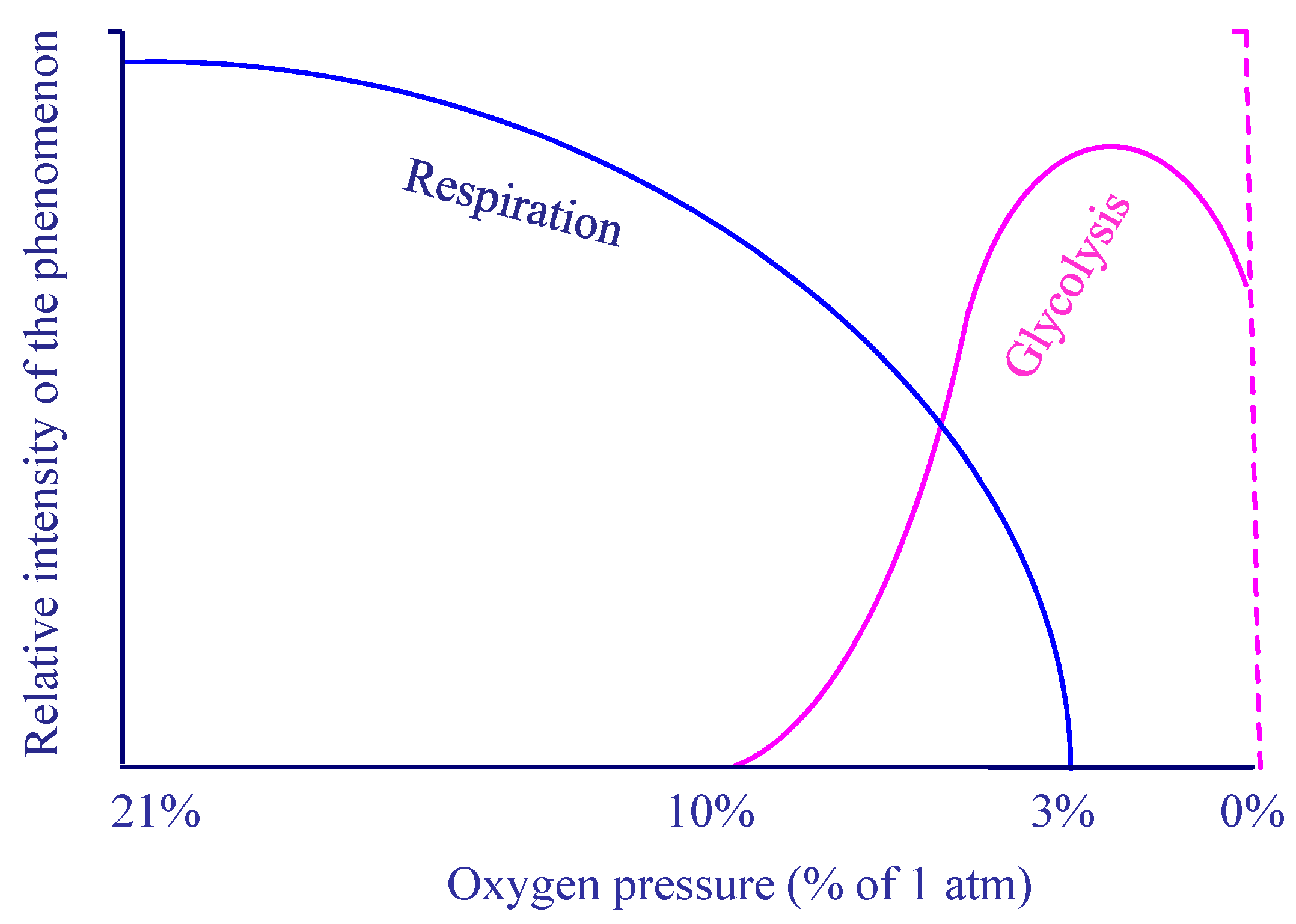
8. Respiration and pH Imbalance
9. Discussion
10. Concluding Remarks
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keibler, M.A.; Wasylenko, T.M.; Kelleher, J.K.; Iliopoulos, O.; Vander Heiden, M.G.; Stephanopoulos, G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, H.; Chun, Y.S.; Lewis, B.C.; Dang, C.V. A unique glucose-dependent apoptotic pathway induced by c366 Myc. Proc. Nat. Acad. Sci. USA 1998, 95, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.H. The Prime Cause and Prevention of Cancer; K. Triltsch: Würtzburg, Germany, 1969; pp. 6–16. [Google Scholar]
- Bose, S.; Le, A. Glucose metabolism in cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [PubMed]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radical Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, D.; Song, Z.L.; Liu, T.; Hou, Y.; Fang, J. Small molecules regulating reactive oxygen species homeostasis for cancer therapy. Med. Res. Rev. 2021, 41, 342–394. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.J.; Seo, Y.R. An overview of carcinogenic heavy metal: Molecular toxicity mechanism and prevention. J. Cancer Prev. 2015, 20, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, A.; Lyndin, M.; Sikora, V.; Lyndina, Y.; Romaniuk, S.; Sikora, K. Heavy metals effect on breast cancer progression. J. Occup. Med. Toxicol. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hemat. 2004, 51, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Blériot, C.; Currenti, J.; Ginhoux, F.; Ginhoux, F. Oncofetal reprogramming in tumour development and progression. Nat. Rev. Cancer 2022, 22, 593–602. [Google Scholar] [CrossRef]
- Lesuffleur, T.; Barbat, A.; Dussaulx, E.; Zweibaum, A. Growth adaptation to methotrexate of HT-29 human colon carcinoma cells is associated with their ability to differentiate into columnar absorptive and mucus-secreting cells. Cancer Res. 1990, 50, 6334–6343. [Google Scholar] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [Green Version]
- Sell, S. (Ed.) Cancer Markers: Diagnostic and Developmental Significance; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 1, pp. 6–18. [Google Scholar] [CrossRef]
- Eales, K.; Hollinshead, K.; Tennant, D. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [Green Version]
- Della Rocca, Y.; Fonticoli, L.; Rajan, T.S.; Trubiani, O.; Caputi, S.; Diomede, F.; Pizzicannella, J.; Guya Diletta Marconi, G.D. Hypoxia: Molecular pathophysiological mechanisms in human diseases. J. Physiol. Biochem. 2022, 78, 739–752. [Google Scholar] [CrossRef]
- Georgakilas, A.G. Oxidative stress, DNA damage and repair in carcinogenesis: Have we established a connection? Cancer Lett. 2012, 327, 3–4. [Google Scholar] [CrossRef]
- Kitao, H.; Iimori, M.; Kataoka, Y.; Wakasa, T.; Tokunaga, E.; Saeki, H.; Oki, E.; Maehara, Y. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018, 109, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Leonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: How, where, and what for? Free Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Banerjee, A.; Banerjee, U.C. Xanthine oxidoreductase: A journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit. Rev. Biotechnol. 2011, 31, 264–280. [Google Scholar] [CrossRef]
- Zhang, Q.; Zou, P.; Zhan, H.; Zhang, M.; Zhang, L.; Ge, R.S.; Huang, Y. Dihydrolipoamide dehydrogenase and cAMP are associated with cadmium-mediated Leydig cell damage. Toxicol. Lett. 2011, 205, 183–189. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.A.D.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signalling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Birsoy, K.; Kong, H.; Martínez-Reyes, I.; Wang, T.; Gao, P.; Sabatini, D.M.; Chandel, N.S. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat. Chem. Biol. 2017, 13, 1274–1279. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.N.; Pierce, J.D.; Clancy, R.L. Reactive oxygen species in acute respiratory distress syndrome. Heart Lung 2001, 30, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Quick, K.L. Reactive oxygen species and aging: Evolving questions. Sci. Aging Knowl. Environ. 2005, 26, pe20. [Google Scholar] [CrossRef]
- Brown, N.S.; Bicknell, R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: Its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001, 3, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, D.; Shinkai, Y.; Kumagai, Y. Signal transduction pathways and transcription factors triggered by arsenic trioxide in leukemia cells. Toxicol. Appl. Pharmacol. 2010, 244, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Pandol, S.J. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology 2004, 4, 567–586. [Google Scholar] [CrossRef]
- Chan, D.W.; Liu, V.W.; Tsao, G.S.; Yao, K.M.; Furukawa, T.; Chan, K.K.; Ngan, H.Y. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008, 29, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: Oxford, UK, 2015; pp. 30–65. [Google Scholar]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, oxidative stress, and metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Sasaki, Y. Does oxidative stress participate in the development of hepatocellular carcinoma. J. Gastroenterol. 2006, 41, 1135–1148. [Google Scholar] [CrossRef]
- Tripathy, B.C.; Oelmüller, R. Reactive oxygen species generation and signaling in plants. Plant Signal. Behav. 2012, 7, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Overmyer, K.; Brosché, M.; Kangasjärvi, J. Reactive oxygen species and hormonal control of cell death. Trends Plant Sci. 2003, 8, 335–342. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative stress and liver cancer: Etiology and therapeutic targets. Oxid. Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Kim, T.; Nakamura, H. Hepatitis, cirrhosis, and hepatoma. J. Magn. Reson. Imaging 1998, 8, 346–358. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Hothorn, T.; Lausen, B.; Benner, A.; Radespiel-Tröger, M. Bagging survival trees. Stat. Med. 2004, 23, 77–91. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Mosley, W.G.; Georgakila, S.; Ziech, D.; Panayiotidis, M.I. Viral-induced human carcinogenesis: An oxidative stress perspective. Mol. BioSystems 2010, 6, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radical. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Lazarus, E.; Bays, H.E. Cancer and obesity: An obesity medicine association (OMA) clinical practice statement (CPS) 2022. Obes. Pillars 2022, 3, 100026. [Google Scholar] [CrossRef]
- Wilcock, D.J.; Badrock, A.P.; Wong, C.W.; Owen, R.; Guerin, M.; Southam, A.D.; Johnston, H.; Telfer, B.A.; Fullwood, P.; Watson, J.; et al. Oxidative stress from DGAT1 oncoprotein inhibition in melanoma suppresses tumor growth when ROS defenses are also breached. Cell Rep. 2022, 39, 110995. [Google Scholar] [CrossRef]
- Rakoczy, K.; Szlasa, W.; Sauer, N.; Saczko, J.; Kulbacka, J. Molecular relation between biological stress and carcinogenesis. Mol. Biol. Rep. 2022, 49, 9929–9945. [Google Scholar] [CrossRef]
- Dalton, S.O.; Boesen, E.H.; Ross, L.; Schapiro, I.R.; Johansen, C. Mind and cancer: Do psychological factors cause cancer? Eur. J. Cancer 2002, 38, 1313–1323. [Google Scholar] [CrossRef]
- Edelman, S.; Kidman, A.D. Mind and cancer: Is there a relationship?—A review of evidence. Aust. Psychol. 1997, 32, 79–85. [Google Scholar] [CrossRef]
- Tosevski, D.L.; Milovancevic, M.P. Stressful life events and physical health. Curr. Opin. Psychiatr. 2006, 19, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, D.; Giese-Davis, J. Depression and cancer: Mechanisms and disease progression. Biol. Psychiatr. 2003, 54, 269–282. [Google Scholar] [CrossRef]
- Tubbs, J.D.; Ding, J.; Baum, L.; Sham, P.C. Immune dysregulation in depression: Evidence from genome-wide association. Brain Behav. Immun. Health 2020, 7, 100108. [Google Scholar] [CrossRef]
- Kisely, S.; Crowe, E.; Lawrence, D. Cancer-related mortality in people with mental illness. JAMA Psychiatr. 2013, 70, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Z.; Wang, Y.X.; Jiang, C.L. Inflammation: The common pathway of stress-related diseases. Front. Hum. Neurosci. 2017, 11, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den stoffwechsel der tumoren. Biochem. Z. 1924, 152, 319–344. [Google Scholar]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Kim, A. Mitochondria in cancer energy metabolism: Culprits or bystanders? Toxicol. Res. 2015, 31, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells. Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Racker, E. Bioenergetics and the problem of tumor growth: An understanding of the mechanism of the generation and control of biological energy may shed light on the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar] [PubMed]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef]
- Birnbaum, M.J.; Haspel, H.C.; Rosen, O.M. Transformation of rat fibroblasts by FSV rapidly increases glucose transporter gene transcription. Science 1987, 235, 1495–1498. [Google Scholar] [CrossRef]
- Dang, C.V.; Hamaker, M.; Sun, P.; Le, A.; Gao, P. Therapeutic targeting of cancer cell metabolism. J. Mol. Med. 2011, 89, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Reinfeld, B.I.; Rathmell, W.K.; Kim, T.K.; Rathmell, J.C. The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell. Mol. Immunol. 2022, 19, 46–58. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadivalagan, C.; Krishnan, A.; Chen, S.J.; Hseu, Y.C.; Muthu, S.; Dhar, R.; Tambuwala, M.M. The Warburg effect in osteoporosis: Cellular signaling and epigenetic regulation of energy metabolic events to targeting the osteocalcin for phenotypic alteration. Cell. Signall. 2022, 100, 110488. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Warburg effect and redox balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef] [PubMed]
- Justus, C.R.; Sanderlin, E.J.; Yang, L.V. Molecular connections between cancer cell metabolism and the tumor microenvironment. Int. J. Mol. Sci. 2015, 16, 11055–11086. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 20, 1926–1936. [Google Scholar] [CrossRef]
- Iheagwam, F.N.; Iheagwam, O.T.; Odiba, J.K.; Ogunlana, O.O.; Chinedu, S.N. Cancer and glucose metabolism: A review on Warburg mechanisms. Trop. J. Nat. Prod. Res. 2022, 6, 661–667. [Google Scholar]
- Zhong, X.; He, X.; Wang, Y.; Hu, Z.; Huang, H.; Zhao, S.; Wei, P.; Li, D. Warburg effect in colorectal cancer: The emerging roles in tumor microenvironment and therapeutic implications. J. Hematol. Oncol. 2022, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Fendt, S. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Pinto, M.M.; Paumard, P.; Bouchez, C.; Ransac, S.; Duvezin-Caubet, S.; Mazat, J.P.; Rigoulet, M.; Devin, A. The Warburg effect and mitochondrial oxidative phosphorylation: Friends or foes. BBA-Bioenergetics 2023, 1864, 148931. [Google Scholar] [CrossRef]
- Pouysségur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Ždralević, M.; Vucetic, M. ‘Warburg effect’ controls tumor growth, bacterial, viral infections and immunity-genetic deconstruction and therapeutic perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef]
- Dienel, G.A.; Cruz, N.F. Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 2016, 138, 14–52. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Dongsung Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 2014, 3, e03342. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavov, N.; Budnik, B.A.; Schwab, D.; Airoldi, E.M.; van Oudenaarden, A. Constant growth rate can be supported by decreasing energy flux and increasing aerobic glycolysis. Cell Rep. 2014, 7, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drochioiu, G. The influence of respiratory and pH imbalance in cancer development. Int. J. Biochem. Res. Rev. 2014, 4, 386–409. [Google Scholar] [CrossRef]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of metabolic supply and demand: Aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Ann. Rev. Cell Develop. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Fu, J.; Du, J.; Xu, W. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int. J. Biol. Sci. 2020, 16, 1495. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Mullarky, E.; Xu, J.; Robin, A.D.; Huggins, D.J.; Jennings, A.; Noguchi, N.; Olland, A.; Lakshminarasimhan, D.; Miller, M.; Tomita, D.; et al. Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorg. Med. Chem. Lett. 2019, 29, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Manoj, K.M.; Nirusimhan, V.; Parashar, A.; Edward, J.; Gideon, D.A. Murburn precepts for lactic-acidosis, Cori cycle, and Warburg effect: Interactive dynamics of dehydrogenases, protons, and oxygen. J. Cell. Physiol. 2022, 237, 1902–1922. [Google Scholar] [CrossRef] [PubMed]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Ahn, W.S.; Dong, W.; Zhang, Z.; Cantor, J.R.; Sabatini, D.M.; Iliopoulos, O.; Stephanopoulos, G. Glyceraldehyde 3-phosphate dehydrogenase modulates nonoxidative pentose phosphate pathway to provide anabolic precursors in hypoxic tumor cells. AIChE J. 2018, 64, 4289–4296. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Zam, W.; Ahmed, I.; Yousef, H. The Warburg effect on cancer cells survival: The role of sugar starvation in cancer therapy. Curr. Rev. Clin. Experim. Pharmacol. 2021, 16, 30–38. [Google Scholar]
- Hermsen, R.; Okano, H.; You, C.; Werner, N.; Hwa, T. A growth-rate composition formula for the growth of E. coli on co-utilized carbon substrates. Mol. Syst. Biol. 2015, 11, 801. [Google Scholar] [CrossRef]
- Hui, S.; Silverman, J.M.; Chen, S.S.; Erickson, D.W.; Basan, M.; Wang, J.; Hwa, T.; Williamson, J.R. Quantitative proteomic analysis reveals a simple strategy of global resource allocation in bacteria. Mol. Syst. Biol. 2015, 11, 784. [Google Scholar] [CrossRef]
- Molenaar, D.; Van Berlo, R.; De Ridder, D.; Teusink, B. Shifts in growth strategies reflect tradeoffs in cellular economics. Mol. Syst. Biol. 2009, 5, 323. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, R.A.; Gawlinski, E.T. A reaction-diffusion model of cancer invasion. Cancer Res. 1996, 56, 5745–5753. [Google Scholar]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Acker, H.H.; Ma, S.; Scolaro, T.; Kaech, S.M.; Mazzone, M. How metabolism bridles cytotoxic CD8+ T cells through epigenetic modifications. Trends Immunol. 2021, 42, 401–417. [Google Scholar] [CrossRef]
- Zhang, L.; Romero, P. Metabolic control of CD8+ T cell fate decisions and antitumor immunity. Trends Mol. Med. 2018, 24, 30–48. [Google Scholar] [CrossRef]
- Lee, N.; Zakka, L.R.; Mihm, M.C., Jr.; Schatton, T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016, 48, 177–187. [Google Scholar] [CrossRef]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L., Jr.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8+ T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.C.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Hongo, R.; Ye, X.; Young, K.; Carbonell, K.; Healey, D.C.C.; Siska, P.J.; Barone, S.; Roe, C.E.; Smith, C.C.; et al. CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 2020, 5, e138729. [Google Scholar] [CrossRef]
- Beezhold, K.; Byersdorfer, C.A. Targeting immuno-metabolism to improve anti-cancer therapies. Cancer Lett. 2018, 414, 127–135. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Ueno, H.; Fay, J.; Banchereau, J. Dendritic cells and immunity against cancer. J. Intern. Med. 2011, 269, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Restifo, N.P. TH17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 2010, 10, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Chih, C.P.; Lipton, P.; Roberts, E.L., Jr. Do active cerebral neurons really use lactate rather than glucose. Trends Neurosci. 2001, 24, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte–neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cerebr. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-García, C.M.; Yellen, G. Neurons rely on glucose rather than astrocytic lactate during stimulation. J. Neurosci. Res. 2019, 97, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Lelièvre, P.; Sancey, L.; Coll, J.L.; Deniaud, A.; Busser, B. The multifaceted roles of copper in cancer: A trace metal element with dysregulated metabolism, but also a target or a bullet for therapy. Cancers 2020, 12, 3594. [Google Scholar] [CrossRef]
- Grubman, A.; White, A.R. Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med. 2014, 16, e11. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Met. Integr. Biometal Sci. 2017, 9, 1501–1512. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef]
- Zischka, H.; Einer, C. Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol. 2018, 102, 71–75. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Polishchuk, R.S. The emerging role of lysosomes in copper homeostasis. Met. Integr. Biometal Sci. 2016, 8, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.D.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP Proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, M.; Lang, J. The roles of metallothioneins in carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Héja, L.; Simon, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.T.; Bourassa, D.; Harankhedkar, S.; McCallum, A.M.; Zlatic, S.A.; Calvo, J.S.; Meloni, G.; Faundez, V.; Fahrni, C.J. Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells. Proc. Natl. Acad. Sci. USA 2019, 116, 12167–12172. [Google Scholar] [CrossRef] [Green Version]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 2013, 304, C768–C779. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.H.; Chen, P.C.; Yeh, M.S.; Hsiung, D.Y.; Wang, C.L. Cu/Zn ratios are associated with nutritional status, oxidative stress, inflammation, and immune abnormalities in patients on peritoneal dialysis. Clin. Biochem. 2011, 44, 275–280. [Google Scholar] [CrossRef]
- Mezzetti, A.; Pierdomenico, S.D.; Costantini, F.; Romano, F.; De Cesare, D.; Cuccurullo, F.; Imbastaro, T.; Riario-Sforza, G.; Di Giacomo, F.; Zuliani, G. Copper/zinc ratio and systemic oxidant load: Effect of aging and aging-related degenerative diseases. Free Radic. Biol. Med. 1998, 25, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Huang, S. Zinc and copper levels in bladder cancer: A systematic review and meta-analysis. Biol. Trace Elem. Res. 2013, 153, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.A.K.; Adly, H.M.; Abdelkhaliq, A.A.; Nassir, A.M. Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr. Urol. 2020, 14, 44–49. [Google Scholar] [CrossRef]
- Yücel, I.; Arpaci, F.; Özet, A.; Döner, B.; Karayilanoglu, T.; Sayar, A.; Berk, Ö. Serum copper and zinc levels and copper/zinc ratio in patients with breast cancer. Biol. Trace Elem. Res. 1994, 40, 31–38. [Google Scholar] [CrossRef]
- Khoshdel, Z.; Naghibalhossaini, F.; Abdollahi, K.; Shojaei, S.; Moradi, M.; Malekzadeh, M. Serum copper and zinc levels among Iranian colorectal cancer patients. Biol. Trace Elem. Res. 2016, 170, 294–299. [Google Scholar] [CrossRef]
- Cunzhi, H.; Jiexian, J.; Xianwen, Z.; Jingang, G.; Shumin, Z.; Lili, D. Serum and tissue levels of six trace elements and copper/zinc ratio in patients with cervical cancer and uterine myoma. Biol. Trace Elem. Res. 2003, 94, 113–122. [Google Scholar] [CrossRef]
- Kucharzewski, M.; Braziewicz, J.; Majewska, U.; Gózdz, S. Selenium, copper, and zinc concentrations in intestinal cancer tissue and in colon and rectum polyps. Biol. Trace Elem. Res. 2003, 92, 1–10. [Google Scholar] [CrossRef]
- Jouybari, L.; Kiani, F.; Islami, F.; Sanagoo, A.; Sayehmiri, F.; Hosnedlova, B.; Do¸sa, M.D.; Kizek, R.; Chirumbolo, S.; Bjørklund, G. Copper concentrations in breast cancer: A systematic review and meta-analysis. Curr. Med. Chem. 2019, 26, 6373–6383. [Google Scholar] [CrossRef]
- Lavilla, I.; Costas, M.; Miguel, P.S.; Millos, J.; Bendicho, C. Elemental fingerprinting of tumorous and adjacent non-tumorous tissues from patients with colorectal cancer using ICP-MS, ICP-OES and chemometric analysis. BioMetals 2009, 22, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, D.; Ikeda, Y.; Nakazawa, S. Quantitative analysis of copper, zinc and copper/zinc ratio in selected human brain tumors. J. Neurooncol. 1993, 16, 109–115. [Google Scholar] [CrossRef]
- Callejón-Leblic, B.; Gómez-Ariza, J.L.; Pereira-Vega, A.; García-Barrera, T. Metal dyshomeostasis based biomarkers of lung cancer using human biofluids. Metallomics 2018, 10, 1444–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blockhuys, S.; Malmberg, P.; Wittung-Stafshede, P. Copper distribution in breast cancer cells detected by time-of-flight secondary ion mass spectrometry with delayed extraction methodology. Biointerphases 2018, 13, E412. [Google Scholar] [CrossRef]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The role of copper in drug-resistant murine and human tumors. BioMetals 2009, 22, 377–384. [Google Scholar] [CrossRef]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, A.; Das, A.; Krett, N.L.; Guzman, G.; Thomas, A.; Mancinelli, G.; Bauer, J.; Ushio-Fukai, M.; Fukai, T.; Jung, B. Nuclear translocation of Atox1 potentiates activin A-induced cell migration and colony formation in colon cancer. PLoS ONE 2020, 15, e0227916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Bond, G.J.; Tsang, T.; Posimo, J.M.; Busino, L.; Brady, D.C. Copper chaperone ATOX1 is required for MAPK signaling and growth in BRAF mutation-positive melanoma. Metallomics 2019, 11, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Sudhahar, V.; Youn, S.W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T. Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Ge, G. Lysyl Oxidase, Extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012, 5, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, R.; Polishchuk, R.S. Activity and trafficking of copper-transporting ATPases in tumor development and defense against platinum-based drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef] [Green Version]
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565. [Google Scholar]
- Fymat, A.L. Genetics, epigenetics and cancer. Cancer Ther. Oncol. Int. J. 2017, 4, 555634. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Mukhtar, H. Lifestyle as risk factor for cancer: Evidence from human studies. Cancer Lett. 2010, 293, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzke, V.A.; Kaaks, R.; Kühn, T. Lifestyle and cancer risk. Cancer J. 2015, 21, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef]
- Selye, H. Stress in Health and Disease; Butterworth-Heinemann: Oxford, UK, 2013; pp. 442–450, 869–872. [Google Scholar]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection versus immunopathology. Allergy Asthma Clin. Immunol. 2008, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhou, L.; Zhang, X.; Yang, X.; Niedermann, G.; Xue, J. Psychological distress and eustress in cancer and cancer treatment: Advances and perspectives. Sci. Adv. 2022, 8, eabq7982. [Google Scholar] [CrossRef]
- Kojima, M.; Wakai, K.; Tokudome, S.; Tamakoshi, K.; Toyoshima, H.; Watanabe, Y.; Hayakawa, N.; Suzuki, K.; Hashimoto, S.; Kawado, M.; et al. Perceived psychologic stress and colorectal cancer mortality: Findings from the Japan Collaborative Cohort Study. Psychosom. Med. 2005, 67, 72–77. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Feodorova, Y.N.; Sarafian, V.S. Psychological stress, cellular and molecular mechanisms. Folia Med. 2012, 54, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Sethi, G.; Nair, A.; Ichikawa, H. Nuclear factor-κB: A holy grail in cancer prevention and therapy. Curr. Sign. Transd. Therap. 2006, 1, 25–52. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Wang, J.; Feng, M.; Peng, J.; Du, X.; Chu, H.; Chen, X. The mitochondrial-derived peptide MOTS-c attenuates oxidative stress injury and the inflammatory response of H9c2 cells through the Nrf2/ARE and NF-κB pathways. Cardiovasc. Eng. Techn. 2022, 13, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Oršolić, N.; Mandić, L.; Sadžak, A.; Šegota, S. Anti-oxidative, anti-inflammatory and anti-apoptotic effects of flavonols: Targeting Nrf2, NF-κB and p53 pathways in neurodegeneration. Antioxidants 2021, 10, 1628. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.I.; Jain, P.; Khan, Z.K. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Heo, S.; Kim, D.-W.; Kim, I.-S.; Ahn, D.; Tae, H.-J.; Kim, M.-K.; Park, B.-Y. Ethanol extract of Maclura tricuspidata fruit protects SH-SY5Y neuroblastoma cells against H2O2-induced oxidative damage via inhibiting MAPK and NF-κB signaling. Int. J. Mol. Sci. 2021, 22, 6946. [Google Scholar] [CrossRef]
- Song, C.; Lv, J.; Yu, C.; Zhu, M.; Yu, C.; Guo, Y.; Yang, L.; Chen, Y.; Chen, Z.; Jiang, T.; et al. Adherence to Healthy Lifestyle and Liver cancer in Chinese: A prospective cohort study of 0.5 million people. Br. J. Cancer 2022, 126, 815–821. [Google Scholar] [CrossRef]
- Rivers, D. Lifestyle interventions for cancer survivors. Nat. Rev. Cancer 2022, 22, 130. [Google Scholar] [CrossRef]
- Argiris, A.; Brockstein, B.E.; Haraf, D.J.; Stenson, K.M.; Mittal, B.B.; Kies, M.S.; Rosen, F.R.; Jovanovic, B.; Rosen, F.R.; Jovanovic, B.; et al. Competing causes of death and second primary tumors in patients with locoregionally advanced head and neck cancer treated with chemoradiotherapy. Clin. Cancer Res. 2004, 10, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Friedenreich, C.M.; Yang, L. Concerns remain regarding the association of sitting time and physical activity with cancer survivorship-reply. JAMA Oncol. 2022, 8, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Cathcart-Rake, E.J.; Ruddy, K.J. Evidence-based guidance for breast cancer survivorship. Hematol. Oncol. Clinics 2023, 37, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Maresso, K.C.; Basen-Engquist, K.; Hawk, E. Cancer epidemiology, prevention, and survivorship. In Perioperative Care of the Cancer Patient; Elsevier: Amsterdam, The Netherlands, 2023; pp. 3–14. [Google Scholar]
- Kousar, K.; Ahmad, T.; Naseer, F.; Kakar, S.; Anjum, S. Immune landscape and immunotherapy options in cervical carcinoma. Cancers 2022, 14, 4458. [Google Scholar] [CrossRef]
- Emery, J.; Butow, P.; Lai-Kwon, J.; Nekhlyudov, L.; Rynderman, M.; Jefford, M. Management of common clinical problems experienced by survivors of cancer. Lancet 2022, 399, 1537–1550. [Google Scholar] [CrossRef]
- Hofmann, P. Cancer and exercise: Warburg hypothesis, tumour metabolism and high-intensity anaerobic exercise. Sports 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Notari, L.; Kirton, R.; Mills, D.S. Psycho-behavioural changes in dogs treated with corticosteroids: A clinical behaviour perspective. Animals 2022, 12, 592. [Google Scholar] [CrossRef]
- Eckerling, A.; Ricon-Becker, I.; Sorski, L.; Sandbank, E.; Ben-Eliyahu, S. Stress and cancer: Mechanisms, significance and future directions. Nat. Rev. Cancer 2021, 21, 767–785. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth Nanda, C.; Venkateswaran, S.V.; Patani, N.; Yuneva, M. Defining a metabolic landscape of tumours: Genome meets metabolism. Br. J. Cancer 2020, 122, 136–149. [Google Scholar] [CrossRef]
- Jögi, A. Tumour hypoxia and the hypoxia-inducible transcription factors: Key players in cancer progression and metastasis. In Tumor Cell Metabolism; Springer: Vienna, Austria, 2015; pp. 65–98. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Kumar, H.; Choi, D.K. Hypoxia inducible factor pathway and physiological adaptation: A cell survival pathway. Mediat. Inflamm. 2015, 2015, 584758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them. Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, C.; Di Bartolomeo, S.; Cecconi, F. Autophagy in stem and progenitor cells. Cell. Mol. Life Sci. 2016, 73, 475–496. [Google Scholar] [CrossRef]
- Buzalewicz, I.; Mrozowska, M.; Kmiecik, A.; Kulus, M.; Haczkiewicz-Leśniak, K.; Dzięgiel, P.; Podhorska-Okołów, M.; Zadka, Ł. Quantitative phase imaging detecting the hypoxia-induced patterns in healthy and neoplastic human colonic epithelial cells. Cells 2022, 11, 3599. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Warmoes, M.O.; Locasale, J.W. Organization of enzyme concentration across the metabolic network in cancer cells. PLoS ONE 2015, 10, e0117131. [Google Scholar] [CrossRef]
- Drochioiu, G. Biological systems: A structural-phenomenological approach. In Cybernetics and Systems, Proceedings of the Nineteenth Meeting on Cybernetics and Systems Research, Vienna, Austria, 25–28 March 2008; Trappl, R., Ed.; Austrian Society for Cybernetic Studies: Vienna, Austria, 2008; Volume 1, pp. 203–208. [Google Scholar]
- Swenson, E.R. The many acid–base manifestations and consequences of hypoxia. Curr. Opin. Physiol. 2019, 7, 72–81. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef]
- Aldwin, C.M. Stress, Coping, and Development: An Integrative Perspective; Guilford Press: New York, NY, USA, 2007; pp. 37–54. [Google Scholar]
- Lovallo, W.R. Stress and Health: Biological and Psychological Interactions, 2nd ed.; Sage: Thousand Oaks, CA, USA; London, UK; New Delhi, India, 1997; pp. 11–22. [Google Scholar]
- Stranks, J. (Ed.) Stress at Work; Burlington: Burlington, NJ, USA, 2005; pp. 4–21, 41–44. [Google Scholar]
- Bendelow, G. Health, Emotion and the Body; Bendelow, G., Ed.; Polity Press: Cambridge, UK; Malden, MA, USA, 2009; pp. 80–105. [Google Scholar]
- Spiegel, D. Cancer. In Encyclopedia of Stress; Fink, G., Ed.; Academic Press: Cambridge, MA, USA, 2000; Volume 1, pp. 368–375. [Google Scholar]
- Kraemer, W.J.; Fleck, S.J.; Deschenes, M.R. Exercise Physiology: Integrating Theory and Application; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; pp. 27–66,103–134, 167–196. [Google Scholar]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2008, 409, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Erecińska, M.; Silver, I.A. Tissue oxygen tension and brain sensitivity to hypoxia. Res. Physiol. 2001, 128, 263–276. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklund, L. Carbon dioxide formation and elimination in man. Upsala J. Med. Sci. 1996, 101, 35–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, J.R.; Jákob, A.; Scholz, R. Energy cost of gluconeogenesis in rat liver. Metabolism 1971, 20, 13–26. [Google Scholar] [CrossRef]
- Robin, E.D. Abnormalities of acid-base regulation in chronic pulmonary disease, with special reference to hypercapnia and extracellular alkalosis. N. Engl. J. Med. 1963, 268, 917–922. [Google Scholar] [CrossRef]
- West, J.B. Pulmonary Physiology and Pathophysiology: An Integrated, Case-Based Approach, 2nd ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Baltimore, MD, USA; Philadelphia, PA, USA, 2007; pp. 15–24. [Google Scholar]
- Das, M.; Bouchey, D.M.; Moore, M.J.; Hopkins, D.C.; Nemenoff, R.A.; Stenmark, K.R. Hypoxiainduced proliferative response of vascular adventitial fibroblasts is dependent on G protein-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 2001, 276, 15631–15640. [Google Scholar] [CrossRef] [Green Version]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Boutilier, R.G. Mechanisms of cell survival in hypoxia and hypothermia. J. Exp. Biol. 2001, 204, 3171–3181. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Angiogenesis: Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef]
- Morowitz, H.; Smith, E. Energy flow and the organization of life. Complexity 2007, 13, 51–59. [Google Scholar] [CrossRef]
- Embley, T.M.; Martin, W. Eukaryotic evolution, changes and challenges. Nature 2006, 440, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, C.A. Cellular senescence and cancer treatment. BBA-Rev. Cancer 2007, 1775, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Nyakas, C.; Buwald, B.; Luiten, P.G.M. Hypoxia and brain development. Prog. Neurobiol. 1996, 49, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Hall, M.E. Guyton and Hall Textbook of Medical Physiology e-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Reglin, B.; Secomb, T.W.; Pries, A.R. Structural adaptation of microvessel diameters in response to metabolic stimuli: Where are the oxygen sensors? Am. J. Physiol. 2009, 297, H2206–H2219. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, N.P.; Pao, A.C.; Raphael, K.L. Acid-mediated kidney injury across the spectrum of metabolic acidosis. Adv. Chronic Kidney Dis. 2022, 29, 406–415. [Google Scholar] [CrossRef]
- Fais, S. Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism. J. Intern. Med. 2010, 267, 515–525. [Google Scholar] [CrossRef]
- Bongaerts, G.P.A.; Van Halteren, H.K.; Verhagen, C.A.M.; Wagener, D.T. Cancer cachexia demonstrates the energetic impact of gluconeogenesis in human metabolism. Med. Hypotheses 2006, 67, 1213–1222. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Brooks, G.A. The science and translation of lactate shuttle theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [Green Version]
- Van Hall, G. Lactate kinetics in human tissues at rest and during exercise. Acta Physiol. 2010, 199, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.C.; Rossiter, H.B.; Brooks, G.A.; Gladden, L.B. The anaerobic threshold: 50+ years of controversy. J. Physiol. 2021, 599, 737–767. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.D.; Glew, R.H. Medical biochemistry: Human Metabolism in Health and Disease; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Oxidation of glucose and fatty acids to CO2. In Molecular Cell Biology, 4th ed.; W.H. Freeman & Comp.: New York, NY, USA, 2000. [Google Scholar]
- Freeman, W.H.; Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006, 175, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Singh, U.P. Role of stress in initiation and progression of cancer: An overview. Indian Exp. Soc. Sci. Human. 2018, 12, 54–59. [Google Scholar]
- Sklar, L.S.; Anisman, H. Social stress influences tumor growth. Psychosom. Med. 1980, 42, 347–365. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Update 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. BBA-Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach. Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Jin Shin, K.; Jin Lee, Y.; Ryoul Yang, Y.; Park, S.; Suh, P.G.; Yung Follo, M.; Cocco, L.; Ho Ryu, S.; Ho Ryu, S. Molecular mechanisms underlying psychological stress and cancer. Curr. Pharm. Des. 2016, 22, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Tu, Y.; Long, Z.; Liu, J.; Kong, D.; Peng, J.; Wu, H.; Zheng, G.; Zhao, J.; Chen, Y.; et al. Reactive oxygen species bridge the gap between chronic inflammation and tumor development. Oxid. Med. Cell. Longev. 2022, 2022, 2606928. [Google Scholar] [CrossRef] [PubMed]
- Drochioiu, G. Chronic metabolic acidosis may be the cause of cachexia: Body fluid pH correction may be an effective therapy. Med. Hypotheses 2008, 70, 1167–1173. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, N. Cancer cachexia and targeting chronic inflammation: A unified approach to cancer treatment and palliative/supportive care. J. Support. Oncol. 2007, 5, 157–162. [Google Scholar]
- Melstrom, L.G.; Melstrom, K.A., Jr.; Ding, X.Z.; Adrian, T.E. Mechanisms of skeletal muscle degradation and its therapy in cancer cachexia. Histol. Histopathol. 2007, 22, 805–814. [Google Scholar]
- Horie, K.; Matsuda, T.; Yamashita, K.; Hasegawa, H.; Utsumi, M.; Urakawa, N.; Kanajia, S.; Oshikiria, T.; Kakeji, Y. Sarcopenia assessed by skeletal muscle mass volume is a prognostic factor for oncological outcomes of rectal cancer patients undergoing neoadjuvant chemoradiotherapy followed by surgery. Eur. J. Surg. Oncol. 2022, 48, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J. European working group on sarcopenia in older people: Sarcopenia: European consensus on definition and diagnosis. Report of the European working group on sarcopenia in older people. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kaur, B.; Singh, P. Inflammation: Biochemistry, cellular targets, anti-inflammatory agents and challenges with special emphasis on cyclooxygenase-2. Bioorg. Chem. 2022, 121, 105663. [Google Scholar] [CrossRef] [PubMed]
- Rittler, P.; Jauch, K.W.; Hartl, W. Metabolische Unterschiede zwischen Anorexie, Katabolie und Kachexie. Akt. Ernähr. Med. 2007, 32, 93–98. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in inflammatory disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Murariu, M.; Drochioiu, G. Biostructural theory of the living systems. BioSystems 2012, 109, 126–132. [Google Scholar] [CrossRef]
- Macovschi, E. The biostructural theory of cancerogenesis. Rev. Roum. Biochim. 1984, 21, 3–11. [Google Scholar]
- Drochioiu, G.; Oniscu, C.; Gradinaru, R.; Murariu, M. The biostructural theory versus the chemiosmotic theory. Rom. Biotechnol. Lett. 2004, 9, 1579–1586. [Google Scholar]
- Drochioiu, G.; Murariu, M. The relationship between the carcinogens in the environment, the biostructure of the living organisms and cancer. Sci. Permits Work. Paper 2001, S1574-0331, 4. [Google Scholar]
- Baiculescu, S.; Acalugaritei, G. (ANs) Networks within the Context of Macovschi’s Biostructural Theory (MBt). In Proceedings of the 12th International WOSC Congress 4th Workshop of IIGSS, Pitssburg, PA, USA, 24–26 March 2002. [Google Scholar]
- Baiculescu, S. Spaces and ideas. In Propedeutics of the Essay “Space of Experience” (Prolegómena); Scholars’ Press: Chisinau, Moldova, 2022; pp. 2–30. [Google Scholar]
- Yang, M.; Darwish, T.; Larraufie, P.; Rimmington, D.; Cimino, I.; Goldspink, D.A.; Jenkins, B.; Koulman, A.; Brighton, C.A.; Ma, M.; et al. Inhibition of mitochondrial function by metformin increases glucose uptake, glycolysis and GDF-15 release from intestinal cells. Sci. Rep. 2021, 11, 2529. [Google Scholar] [CrossRef]
- Hägg, M.; Wennström, S. Activation of hypoxia-induced transcription in normoxia. Exp. Cell Res. 2005, 306, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Martin, D.S.; Xu, R.A.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Haugrud, A.B.; Zhuang, Y.; Coppock, J.D.; Miskimins, W.K. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drochioiu, G. The role of bacteriorhodopsin in light harvesting and ATP production by Halobacterium salinarum cells. Int. Multidiscip. Sci. GeoConf. SGEM 2022, 22, 137–144. [Google Scholar]
- Hoppeler, H.; Vogt, M.; Weibel, E.R.; Flück, M. Response of skeletal muscle mitochondria to hypoxia. Exp. Physiol. 2003, 88, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; van der Windt, D.J.; Huang, H.; Yazdani, H.O.; Liu, Y.; Loughran, P.; van der Windt, D.J.; et al. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Bhujade, A.; Gupta, G.; Talmale, S.; Das, S.K.; Patil, M.B. Induction of apoptosis in A431 skin cancer cells by Cissus quadrangularis Linn stem extract by altering Bax–Bcl-2 ratio, release of cytochrome c from mitochondria and PARP cleavage. Food Funct. 2013, 4, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radical Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drochioiu, G. Multifactorial Distress, the Warburg Effect, and Respiratory and pH Imbalance in Cancer Development. Stresses 2023, 3, 500-528. https://doi.org/10.3390/stresses3020036
Drochioiu G. Multifactorial Distress, the Warburg Effect, and Respiratory and pH Imbalance in Cancer Development. Stresses. 2023; 3(2):500-528. https://doi.org/10.3390/stresses3020036
Chicago/Turabian StyleDrochioiu, Gabi. 2023. "Multifactorial Distress, the Warburg Effect, and Respiratory and pH Imbalance in Cancer Development" Stresses 3, no. 2: 500-528. https://doi.org/10.3390/stresses3020036