
Ageladine A, a Bromopyrrole Alkaloid from the Marine Sponge Agelas nakamurai


National Hellenic Research Foundation, Institute of Chemical Biology, 48 Vassileos Constantinou Avenue, 11653 Athens, Greece
Compounds 2023, 3(1), 107-121; https://doi.org/10.3390/compounds3010010
Submission received: 4 January 2023
/
Revised: 15 January 2023
/
Accepted: 17 January 2023
/
Published: 19 January 2023
(This article belongs to the Special Issue Feature Papers in Compounds (2022–2023))
Abstract
:During the last three decades, secondary metabolites of marine origin have emerged as a significant source of bioactive compounds. Among the marine organisms explored, sponges offer a vast number of metabolites with unique structural diversity and a plethora of biological activities. Ageladine A, a fluorescent bromopyrrole alkaloid isolated from the marine sponge Agelas nakamurai, exhibited matrix metalloproteinase (MMP) inhibitory properties, as well as antiangiogenic activity. Due to this interesting biological profile, Ageladine A became, soon after its discovery, a target for total synthesis. In addition, a significant number of derivatives have been synthesized, and their biological activity was evaluated. The present review highlights all the successful efforts made towards the synthesis of Ageladine A. Furthermore, all the medicinal chemistry approaches to identify and assess new more potent inhibitors and to elucidate the structural features responsible for the activity are described.
1. Introduction
Nature is considered an unlimited source of chemical compounds with significant biological activities. Long before the development of new methodologies for the discovery of synthetic drugs and the revolution of modern medicine, our ancestors clearly had the notion that nature could provide them with some of the necessary weapons to fight diseases. Despite the fact that more than 70% of our planet is covered by oceans, terrestrial plants and bacteria have always been the primary sources of natural products. However, during the last 30 years, researchers have raised their attention to marine natural products (MNPs) in terms of isolation, structural elucidation, synthesis, and biological evaluation, with bryozoans, microorganisms, sponges, molluscs, tunicates, and algae being, among others, the major sources of MNPs. Of particular importance, a large number of secondary metabolites discovered in marine organisms are derived from marine sponges [1].
Secondary metabolites isolated from marine sponges of the genus Agelas (class Demospongiae, order Agelasida, family Agelasidae) display an amazing structural diversity of bromopyrrole and diterpene alkaloids [2]. To this date, 36 species of the Agelas genus have been identified [3]. Structurally, pyrrole alkaloids generally have a bromo- or dibromopyrrole-2-carboxamide core attached to a variety of side chains, either linear or cyclic [4,5]. Apart from their structural diversity, this class of compounds has shown a wide range of biological activity, including antiparasitic and antimalarial [6,7], antihistaminic [8], antimicrobial [9], antifouling [10], and inhibitory action towards voltage-gated K+ channels [11].
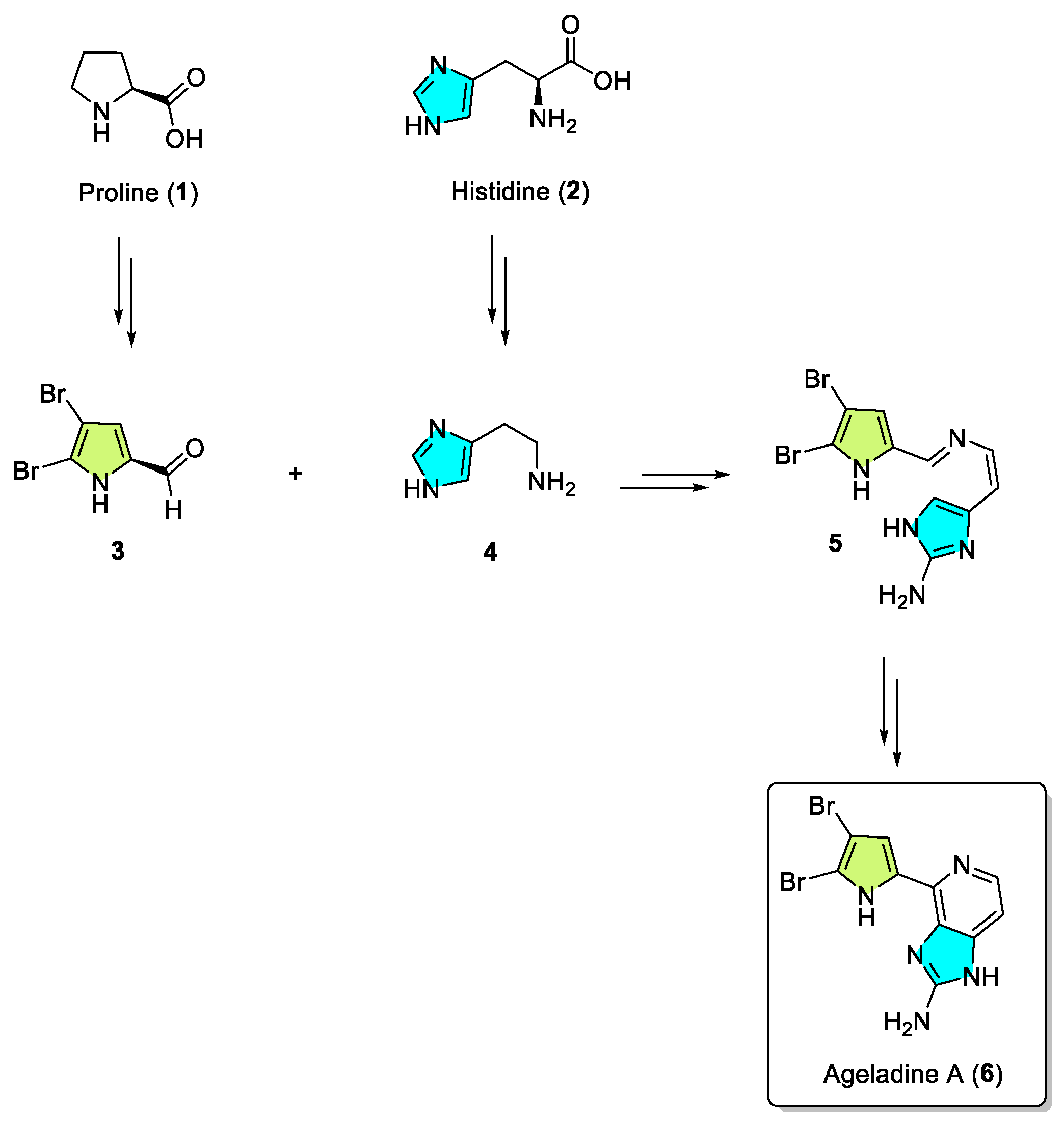
This review primarily deals with the synthetic approaches of the bromopyrrole alkaloid Ageladine A (1) isolated from the marine sponge Agelas nakamurai (Figure 1). Ageladine A was first reported in 2003 by Fusetani et al. [12]. It was isolated from the hydrophilic extract of the marine sponge Agelas namamurai using a bioassay-guided fractionation. Ageladine A belongs to the pyrrole-imidazole alkaloids family, as it was confirmed by two-dimensional (2D) NMR studies. This alkaloid showed inhibition of matrix metalloproteinases MMP-1, -2, -8, -9, -12, and -13 and inhibition of cell migration of bovine aortic endothelial (BAE) cells. Interestingly, kinetic studies revealed that Ageladine A is not able to chelate Zn2+, which is a common feature for MMP-2 inhibitors. Hence, its inhibitory activity is probably attributed to a mechanism different from what other MMP-2 inhibitors exhibited. Previous experiments in the literature, involving radio-labeled amino acids, have shown that active metabolites of the oroidin family can be derived from biogenetic precursors, such as proline, histidine, or ornithine [13,14,15]. Since Ageladine A is an oroidin metabolite, Fusetani et al. proposed a plausible biosynthetic pathway, which is depicted in Figure 1. It is assumed that 4,5-dibromo-1H-pyrrole-2-carbaldehyde (3) can be derived from proline (1), while histamine (4) can be obtained from histidine (2). Then, 2 and 3 can produce N-vinyl imine (5). Finally, this azatriene is subjected to an intramolecular 6π-azaelectrocyclization followed by dehydrogenation of the anticipated dihydropyridine to afford Ageladine A.
Apart from this biological profile, Ageladine A exhibits a pH-dependent fluorescence [16]. It shows the highest fluorescence at pH 3–4, as well as the lowest at pH 9, and the largest fluorescence changes were observed at the region of pH 6–7. Furthermore, due to its membrane permeability, Ageladine A is an excellent candidate for the detection of intracellular pH changes. Hence, it can be used for the staining of living cells. Bickmeyer et al. successfully exploited these properties to stain acidic vesicles in mouse neuronal cells, while it showed no toxicity in either PC12 or hippocampal neurons [17]. All these very interesting properties led to the synthesis of a series of derivatives, and their biological activity was assessed. In the context of the present review, these structure-activity relationship studies of Ageladine A and its derivatives are also discussed.
2. Total Syntheses of Ageladine A
2.1. Weinreb’s Total Synthesis of Ageladine A
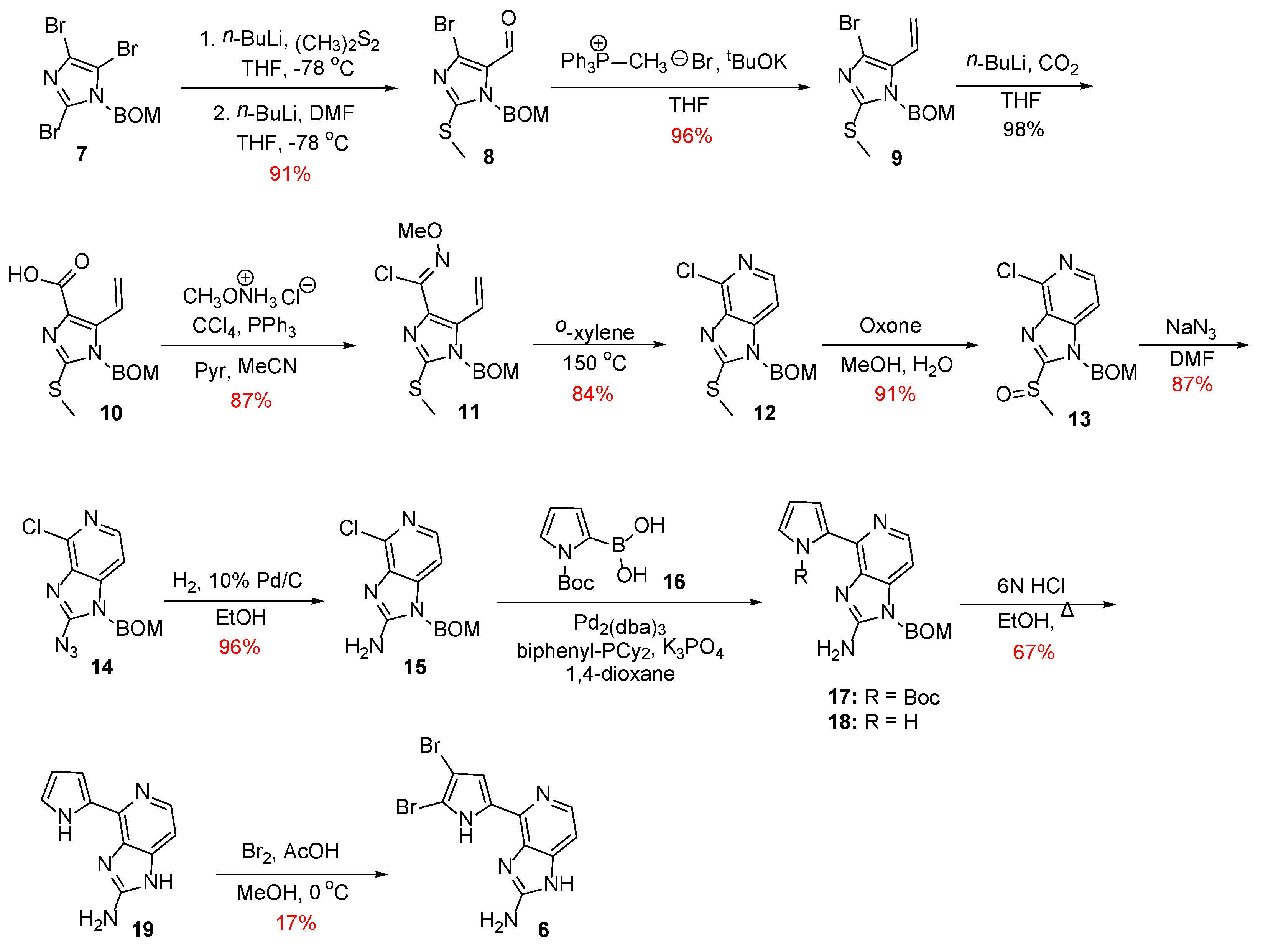
In 2006, Meketa and Weinreb reported the first total synthesis of Ageladine A in 12 steps, using a 6π-azaelectrocyclization approach for the formation of the pyridine ring [18]. This successive strategy is applied in the synthesis of various natural products and their analogs. It involves the conversion of three π-bonds of an hexatriene into a cyclic moiety that contains two π-bonds and a new σ-bond of lower energy [19]. As shown in Scheme 1, the synthesis commenced with the metalation of BOM-protected tribromoimidazole 7 [20], a process that can be realized in a controlled and predictable manner [21,22]. This involved initially treatment of compound 7 with n-BuLi to result in the metalation at the C-2 position followed by the insertion of a thiomethyl group upon reaction with dimethyl disulfide. Then, a second equivalent of n-BuLi was added to effect metalation at the C-5 position and then treated with DMF to introduce a formyl group. Thus, compound 7 was converted to compound 8 through a one-pot procedure.
Towards the formation of the pyridine ring, aldehyde 8 was converted to its corresponding terminal alkene 9 through a Wittig reaction. On the other hand, the bromo substituent was converted to the carboxylic acid 10 upon treatment with n-BuLi and CO2, which in turn gave rise to N-methoxy imidoyl chloride 11 in 87% yield. Finally, a 6π-electrocyclization reaction took place when compound 11 was heated at 150 °C using a solvent of high boiling point, such as o-xylene, to afford 13 in 84% yield.
The next steps on the synthesis of Ageladine A involved the installation of the amino-group at the C-2 position of the imidazole core. To this end, the thiomethyl group was oxidized to the corresponding sulfoxide 13, which then treated with sodium azide to give azide 14. Finally, compound 14 was subjected to catalytic hydrogenation to afford 2-aminoimidazolopyridine 15 with very good overall yield.
The attachment of the pyrrole moiety to intermediate 15 proved to be quite challenging. However, the authors found that this could be realized under Suzuki-Miyaura coupling conditions using as ligand, Buchwald’s 2-biphenyldicyclohexylphosphine [23]. It is worth to notice that this ligand was crucial for the successful outcome of the reaction, since the use of other ligands did not afford the desired product. Thus, compound 15 was coupled with boronic acid 16 to give a mixture of compounds 17 and 18, which was further treated with HCl to result in compound 19 after global deprotection. However, late-stage bromination of pyrrole was problematic. The best results were obtained when Ageladine A was treated with bromine in a cold mixture of acetic acid/methanol. Thus, the desired alkaloid was obtained, but only in 17% yield along with recovered starting material, mono-brominated, and traces of tri-brominated product.
2.2. Karuso’s Total Synthesis of Ageladine A
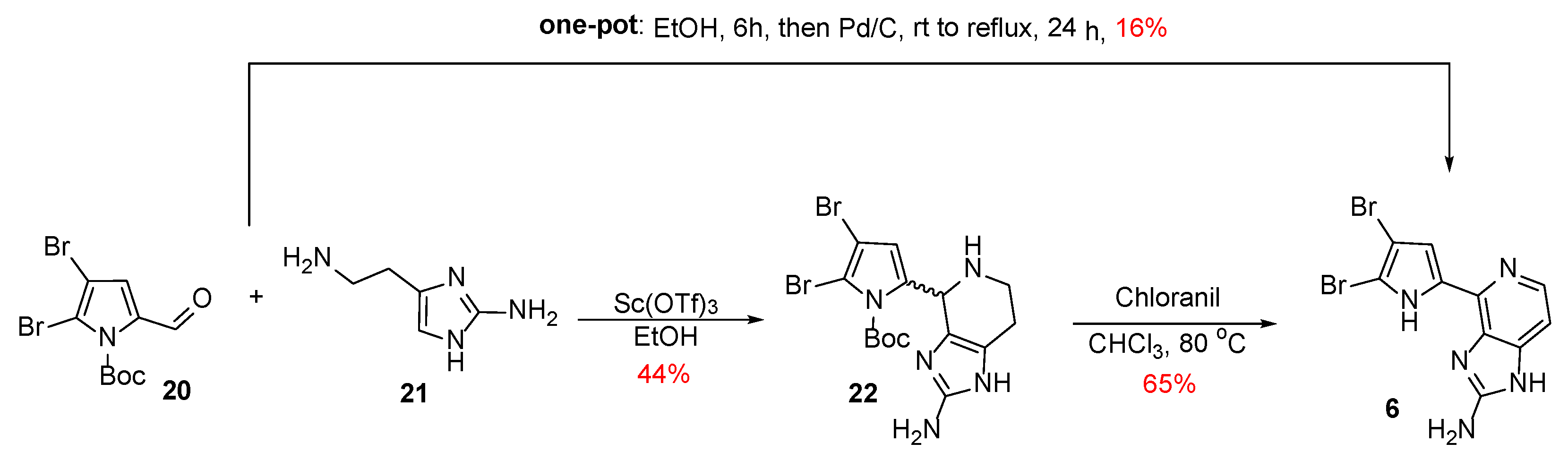
Karuso et al. chose a convergent methodology for the synthesis of Ageladine A using a biomimetic approach [24]. Furthermore, the problematic late-stage bromination of Weinreb’s synthesis was taken under consideration and for that reason, N-Boc-4,5-dibromo-2-formylpyrrole 20 was used as starting material (Scheme 2). According to the proposed biogenesis of Ageladine A [12], it is quite possible that this dibrominated pyrrole can be derived from proline (1), while 2-aminohistamine 21 can be derived from histidine (2). Therefore, a Pictet-Spengler reaction of 20 and 21 in the presence of Sc(OTf)3 as a Lewis acid led to the anticipated compound 22 as a mixture of diastereoisomers. Finally, Ageladine A was successfully obtained in 29% yield after dehydrogenation and simultaneous Boc-deprotection when treated with chloranil in refluxing chloroform.
A few years later, Karuso et al. also reported a robust one-pot procedure for the synthesis of Ageladine A [25]. This procedure involved the Pictet-Spengler reaction of compounds 20 and 21 in the absence of a Lewis acid in ethanol for 6 h while chloranil was replaced by Pd/C (Scheme 2). The resulting mixture was refluxed for 24 h and furnished upon purification Ageladine A in 16% overall yield.
2.3. Ando’s Total Synthesis of Ageladine A
At the time first Karuso’s total synthesis was published, Ando et al. proposed the synthesis of Ageladine A following the same biosynthetic route [26]. However, Ando used Boc-protected aminohistamine 24, instead of Karuso’s completely unprotected 21, which was derived from 23 through a two-step procedure, which involved a nitroaldol condensation with AcONH4 and CH3NO2 followed by reduction with LiAlH4 (Scheme 3). Compound 24 was then subjected to a Pictet-Spengler reaction with SEM-protected 4,5-dibromo-2-formylpyrrole 25 to provide compound 26 in 80% yield. It should be noted that compound 26 was obtained in higher yield without the use of a Lewis acid, compared to Karuso’s synthesis.
For the formation of the pyridine ring (compound 27), Ando et al. followed a two-step procedure. Initially, partial dehydrogenation took place upon treatment of 26 with IBX in DMSO. The dehydrogenation was completed after treatment of the thus obtained intermediate with activated MnO2. The overall yield of these two steps was 89% and proved to be more efficient compared to Karuso’s procedure using chloranil. Finally, Ageladine A was obtained as its bistrifluoroacetate salt after treatment with BF3∙Et2O to remove both protective groups and TFA in MeOH in 70% yield over two steps.
2.4. Weinreb’s Second Total Synthesis of Ageladine A
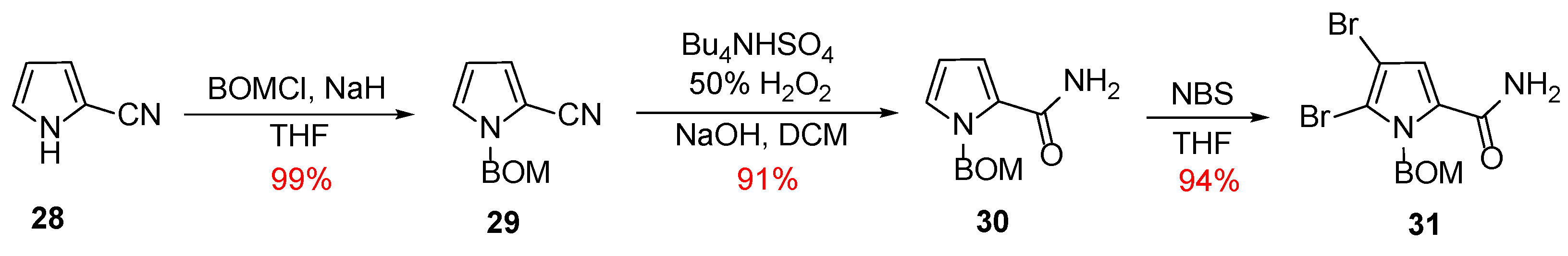
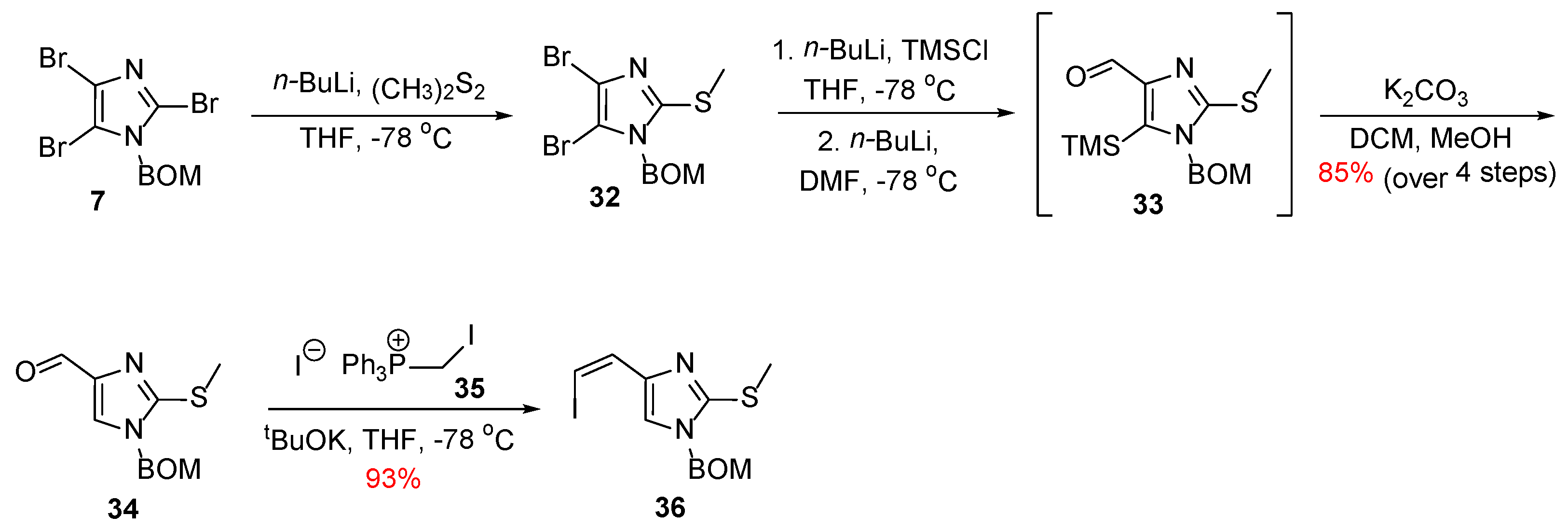
In 2007, Weinreb published a new convergent synthesis of Ageladine A using a biomimetically inspired 6π-2-azatriene electrocyclization for the generation of the imidazolopyridine moiety [27,28]. In contrast with his first synthesis, Weinreb chose to introduce the two bromo-substituents from the beginning of the synthesis. Hence, the necessary pyrrole fragment was synthesized from the commercially available 2-cyanopyrrole 28 as it can be depicted in Scheme 4. Initially, 28 was protected almost quantitively as its corresponding N-BOM derivative (compound 29). Compound 29 was subjected to hydrolysis to give the corresponding primary amide 30, which, upon treatment with NBS, afforded the dibromo derivative 31 in 94% yield with the desired regiochemistry as confirmed by X-ray crystallography.
The construction of the imidazolopyridine fragment started with tribromoimidazole 7, which was transmetallated selectively at the C-2 position to insert a thiomethyl group. Following a procedure already described in their previous synthesis (Scheme 1), compound 34 was obtained, which was then subjected to a Wittig reaction with phosphonium iodide 35 to give vinyl iodide 36 in 93% yield (Scheme 5).
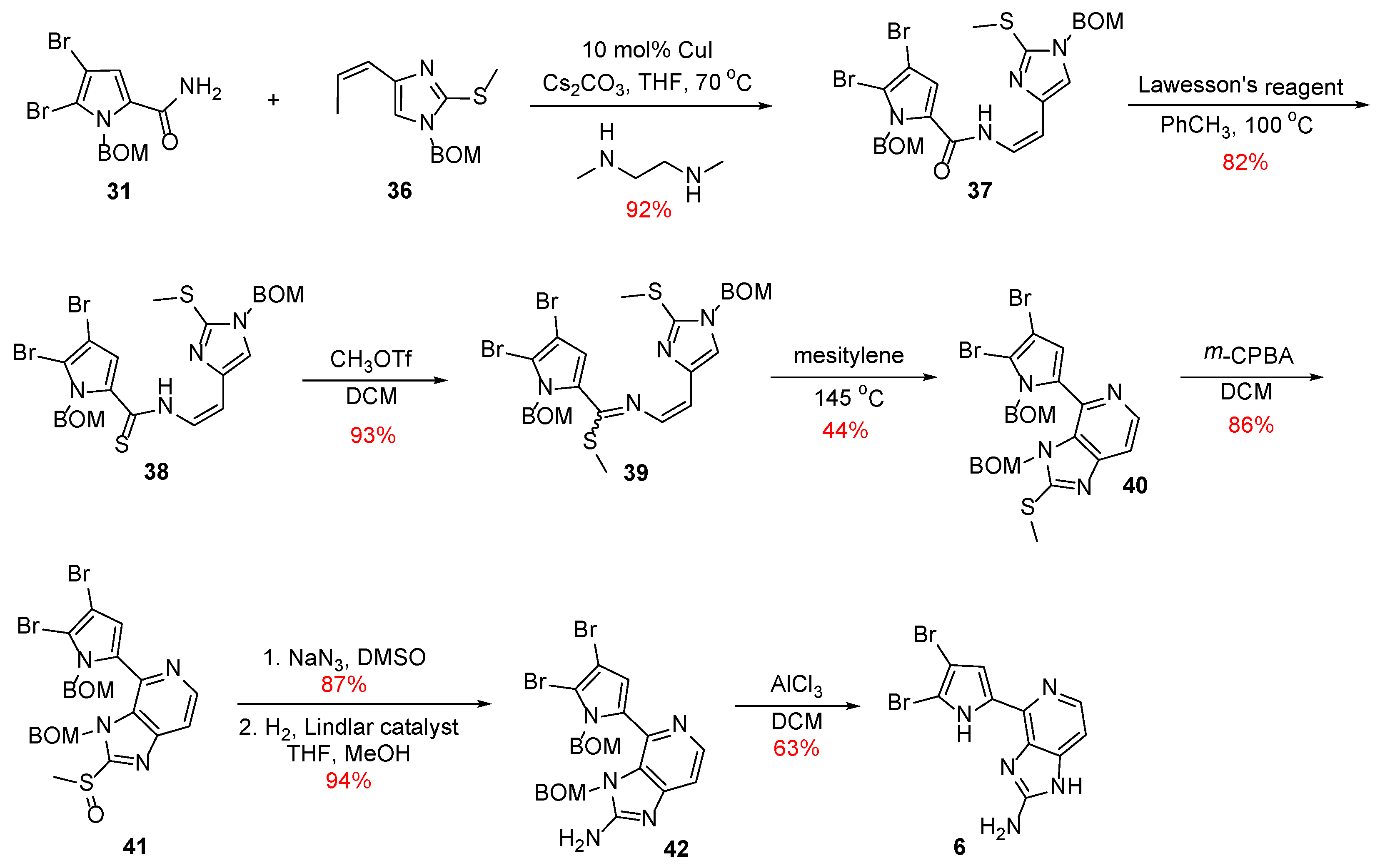
With both fragments in hand, Z-enamide 37 was synthesized stereoselectively using Buchwald’s protocol (Scheme 6) [29], which, in turn, was then converted to its corresponding thioenamide 38 upon treatment with Lawesson’s reagent. Compound 38 was reacted with methyl triflate, and the thiomethyl imidate 39 was formed, which was heated with mesitylene to result in compound 40 in the context of the electrocyclization step. The total synthesis of Ageladine A was completed with the conversion of the thiomethyl group to the corresponding amino function with a procedure described previously.
2.5. Tanaka’s Total Synthesis of Ageladine A
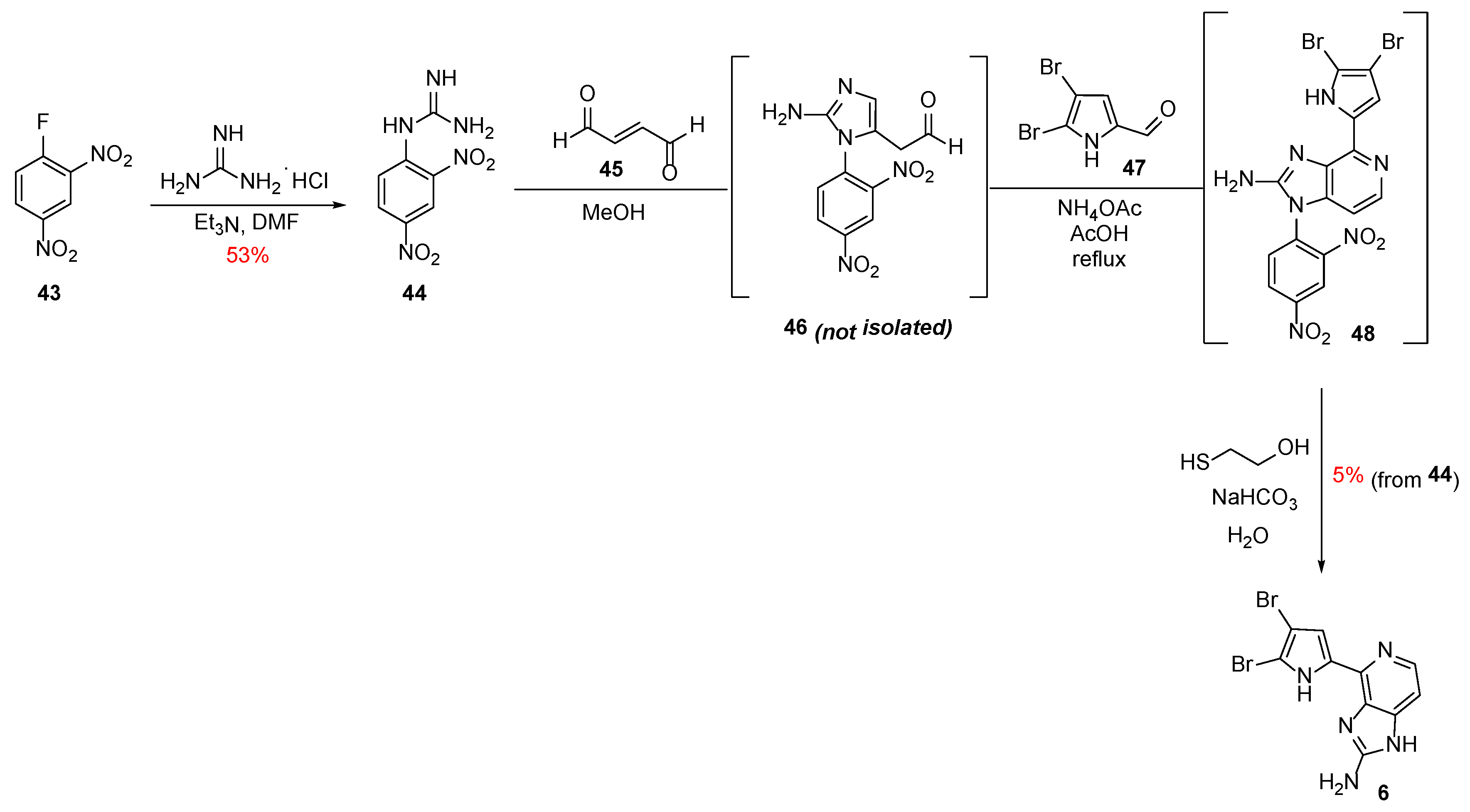
In 2016, Tanaka et al. reported the one-pot synthesis of a new class of Ageladine A derivatives with N1-substitution (vide infra) as selective modulators of neuronal differentiation [30,31]. They used a cascade which involved a new preparation of 2-aminoimidazole inspired by the naturally occurring post-translational modification (PTM) of arginine by lipid metabolites [32]. In proteins, the guanidine moiety of the arginine residue can react with the conjugated aldehyde of the lipid-oxidized metabolite 4-oxo-(2E)-nonenal to give N1-substituted 2-aminoimidazole.
Hence, starting from the commercially available 2,4-dinitrofluorobenzene 43, 2,4-dinitrophenylguanidine 44 was obtained in 53% yield (Scheme 7). This was then reacted with fumaraldehyde 45 to give rise to the intermediate 46, which directly coupled with 4,5-dibromo-2-formylpyrrole 47 to furnish the 2,4-dinitrophenyl-substituted Ageladine A 48. Finally, basic thiolysis of 48 with 2-mercaptoethanol gave rise to Ageladine A in 5% overall yield starting from 44.
2.6. Lindel’s Total Synthesis of Ageladine A
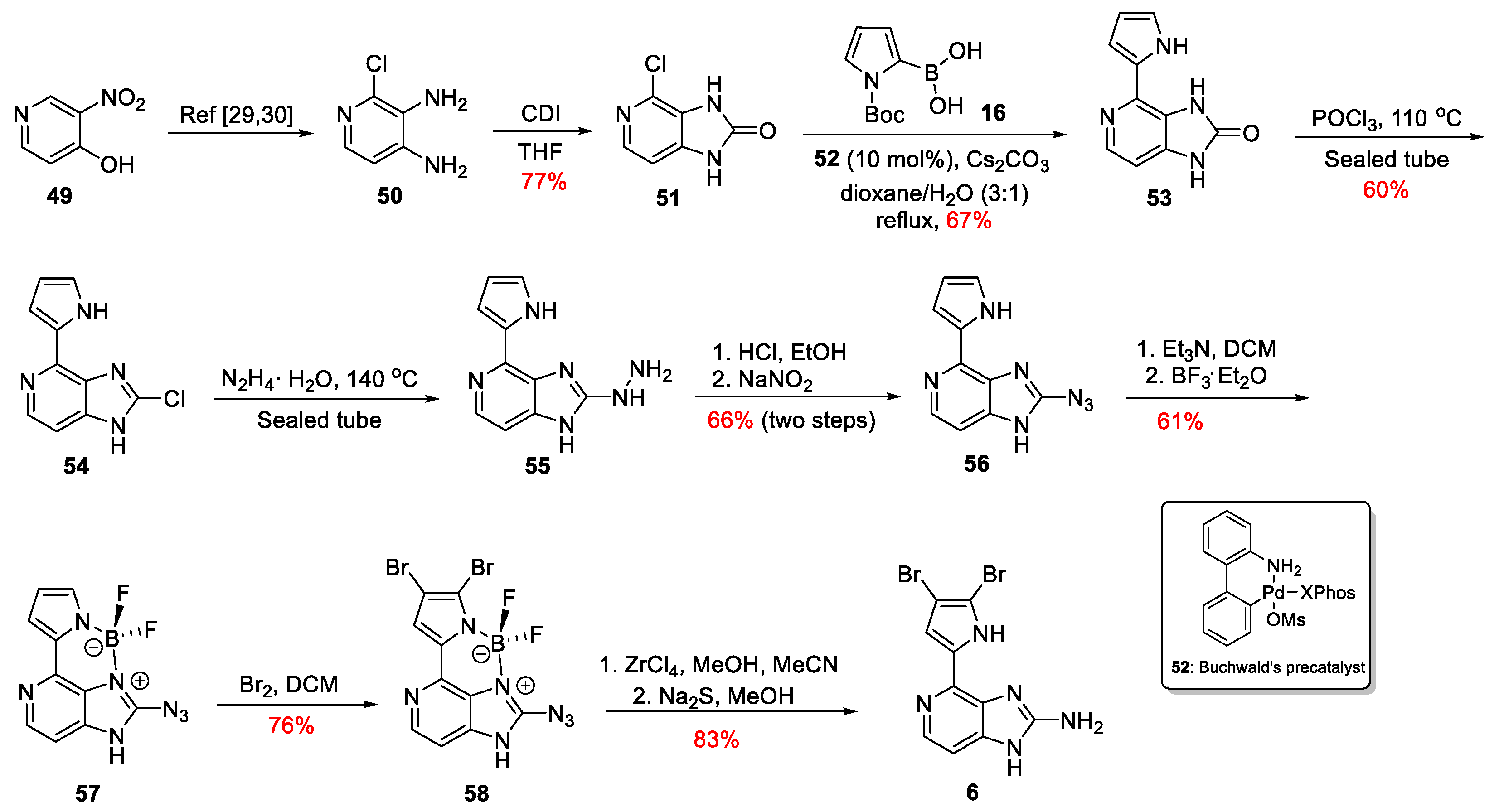
Only recently, Lindel et al. proposed a new alternative for the late-stage bromine insertion to the synthesis of Ageladine A [33]. The regioselective dibromination of the pyrrole ring was accomplished using an aza-BODIPY route (Scheme 8). Initially, 3-nitropyridin-4-ol 49 gave 2-chloropyridine-3,4-diamine 50 through an already published three-step procedure [34,35]. Compound 50 was then treated with CDI to furnish imidazolone 51 in 77% yield. Imidazolone 51 was converted to 53 through a cross-coupling reaction with Boc-protected pyrrole-2-boronic acid 16. The optimum yield for this reaction was obtained when the Buchwald’s precatalyst 52 was used [36]. Treatment of compound 53 with POCl3 in a sealed tube afforded chloride 54, which, upon reaction with hydrazine hydrate and diazotization of the thus obtained compound 55, led to the azide 56 in 66% yield over two steps. Boron complex 57 was then easily obtained upon treatment with BF3∙Et2O in 61% yield, followed by bromination with Br2 to obtain the compound 58 in 76% yield. Hence, the presence of the boron complex directs the insertion of the two bromine atoms to the desired positions. The last two steps towards the synthesis of Ageladine A involved decomplexation with ZrCl4 and finally reduction of the azido function to its corresponding amine using Na2S in MeOH. Thus, Ageladine A was synthesized through nine linear steps in 7.9% overall yield.
3. Synthesis and Biological Evaluation of Ageladine A Derivatives
MMPs are zinc-dependent proteolytic enzymes that can affect cell function through the regulation of membrane receptors’ activity and post-receptor’s signaling mechanisms [37]. Furthermore, they have the ability to degrade various proteins in the extracellular matrix (ECM), such as collagen and elastin and, as a consequence, can interfere in vascular smooth muscle (VSM) cell migration in Ca2+ signaling and proliferation [38]. The role of MMPs in various physiological processes, such as angiogenesis, morphogenesis, or embryogenesis, is well established, while they also play an important role in pathological conditions. Elevated MMPs levels have been associated with progression and invasion of tumors, hence they can be considered as biomarkers for a series of diseases and as appealing therapeutic targets for cardiovascular disorders and cancer.
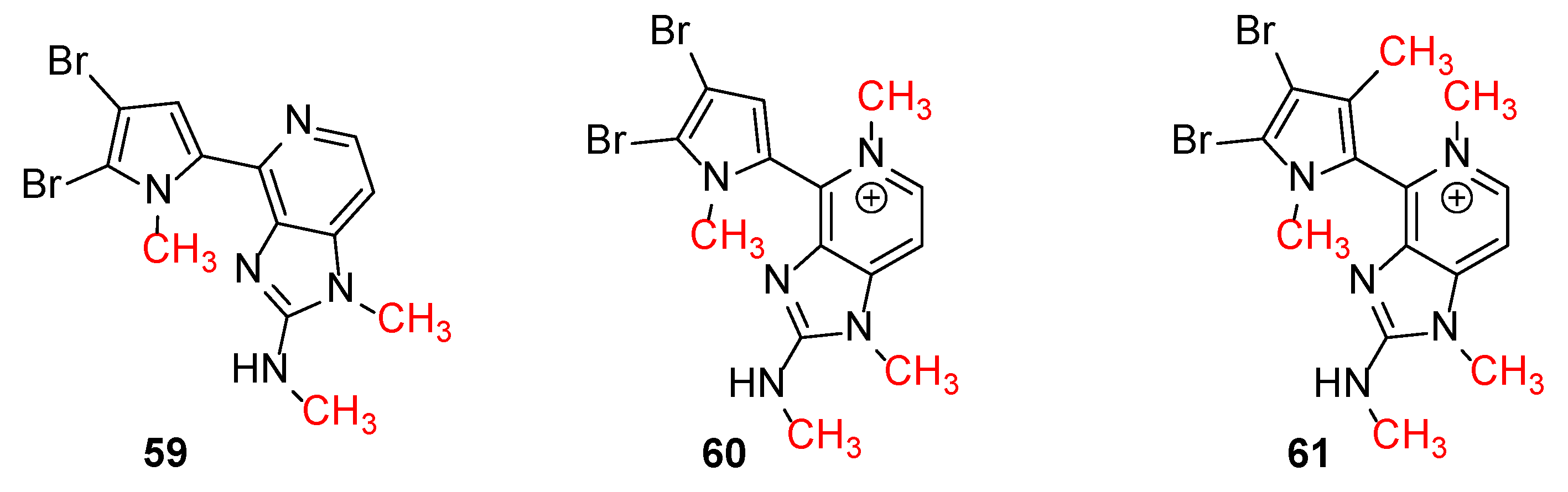
Ageladine A was discovered during Fusetani’s campaign for MMPs inhibitors from Japanese marine invertebrates [12]. It showed inhibitory activity against MMP-2 with IC50 = 2.0 μg/mL but it was also active against other MMPs. Interestingly, the corresponding N-methylated derivatives 59–61 (Figure 2), showed no inhibitory activity against MMP-2. Furthermore, Ageladine A exhibited a 65.9% inhibition of cell migration using BAE cells at 25 μg/mL. This, in combination with the fact that Ageladine A inhibits MMP-2, renders it as a potent antiangiogenic compound.
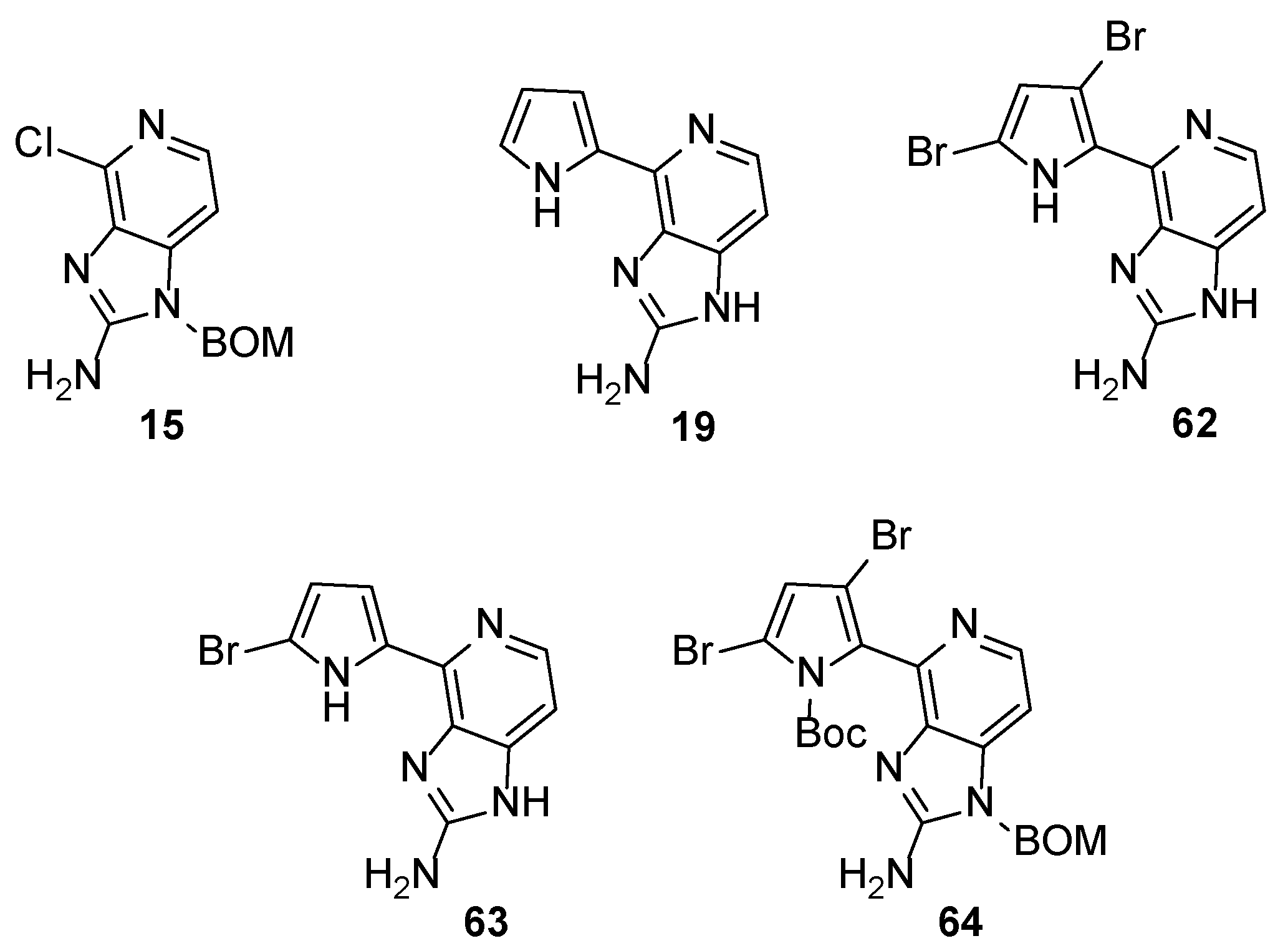
These observations paved the way for the synthesis of new analogues in an effort to find more potent inhibitors and to decipher the structural features responsible for the activity. Weinreb et al. tested some analogues (62–64) and intermediates (15 and 19) derived from the total synthesis of Ageladine A for MMP inhibition (Figure 3) [39].
The screening revealed that the number of bromine atoms, as well as the location, plays vital role to the activity. Brominated compounds 62 and 63 showed superior activity compared to non-brominated compound 19 but were five-fold less active when compared to Ageladine A. Furthermore, the absence of the pyrrole ring (compound 15) or the protection of the nitrogen (compound 64) were detrimental to the activity.
Ando et al. reported the total synthesis of Ageladine A in 2007 [26]. In this paper, they employed their strategy to further synthesize a series of analogues (65–73), which tested as potent MMP-12 inhibitors (Figure 4). This series comprised two mono-brominated analogues (65 and 66), a compound with no bromine (67), compounds 67–72, which are N-methylated in various combinations, and compound 73 lacks the amino function at the C-2 position of the imidazole.
The biological evaluation revealed that the presence of two bromines in the pyrrole ring is indispensable for the inhibition since compounds 65–67 showed no activity (IC50 > 100 μM). The same results were obtained when either pyrrole nitrogen was methylated (compound 68) or imidazole nitrogen was methylated in combination with methylation of the C-2 amino function (compounds 71 and 72). It seems that retention of the two bromines in combination with unsubstituted nitrogen atoms in both pyrrole and imidazole play important role for the activity against MMP-12 since compound 69 was the most active (IC50 = 10.4 μM). However, 69 was three-fold less potent than Ageladine A (IC50 = 3.66 μM).
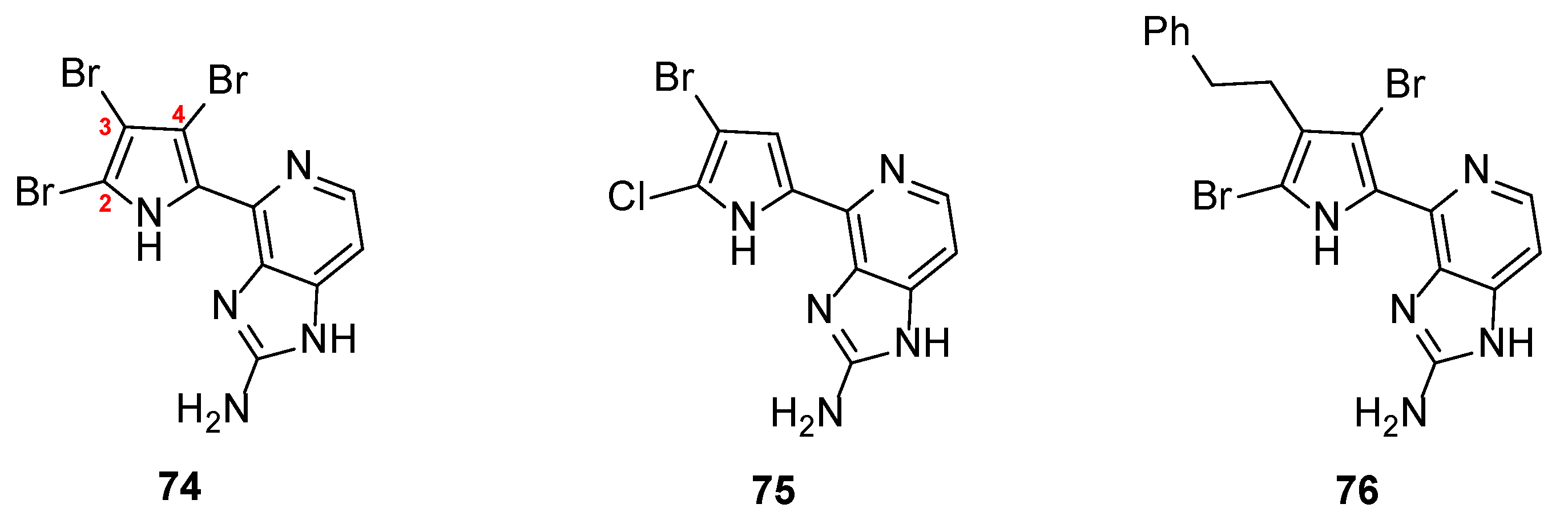
Prompted by these results, Ando et al. reported the synthesis of 21 new analogs in an effort to extent their structure–activity relationship studies (SAR) towards the inhibition of MMP-12. The new compounds bore substituents at the three available positions of the pyrrole ring leaving the imidazopyridine core intact [40]. Rewardingly, three new analogs (74–76) showed better inhibitory activity than Ageladine A itself (Figure 5). The results clearly demonstrated that the presence of a halogen, either bromine or chlorine at the C-2 position of the pyrrole, is crucial for the activity. The substitution at the C-3 position either with bromine or with phenyl or phenethyl groups showed slightly different effect. Interestingly, the introduction of a bromine at the C-4 position significantly increased the activity. Especially, tri-brominated compound 74 exhibited the best activity (IC50 = 1.24 μΜ), being three-fold more active compared to Ageladine A (IC50 = 3.66 μM).
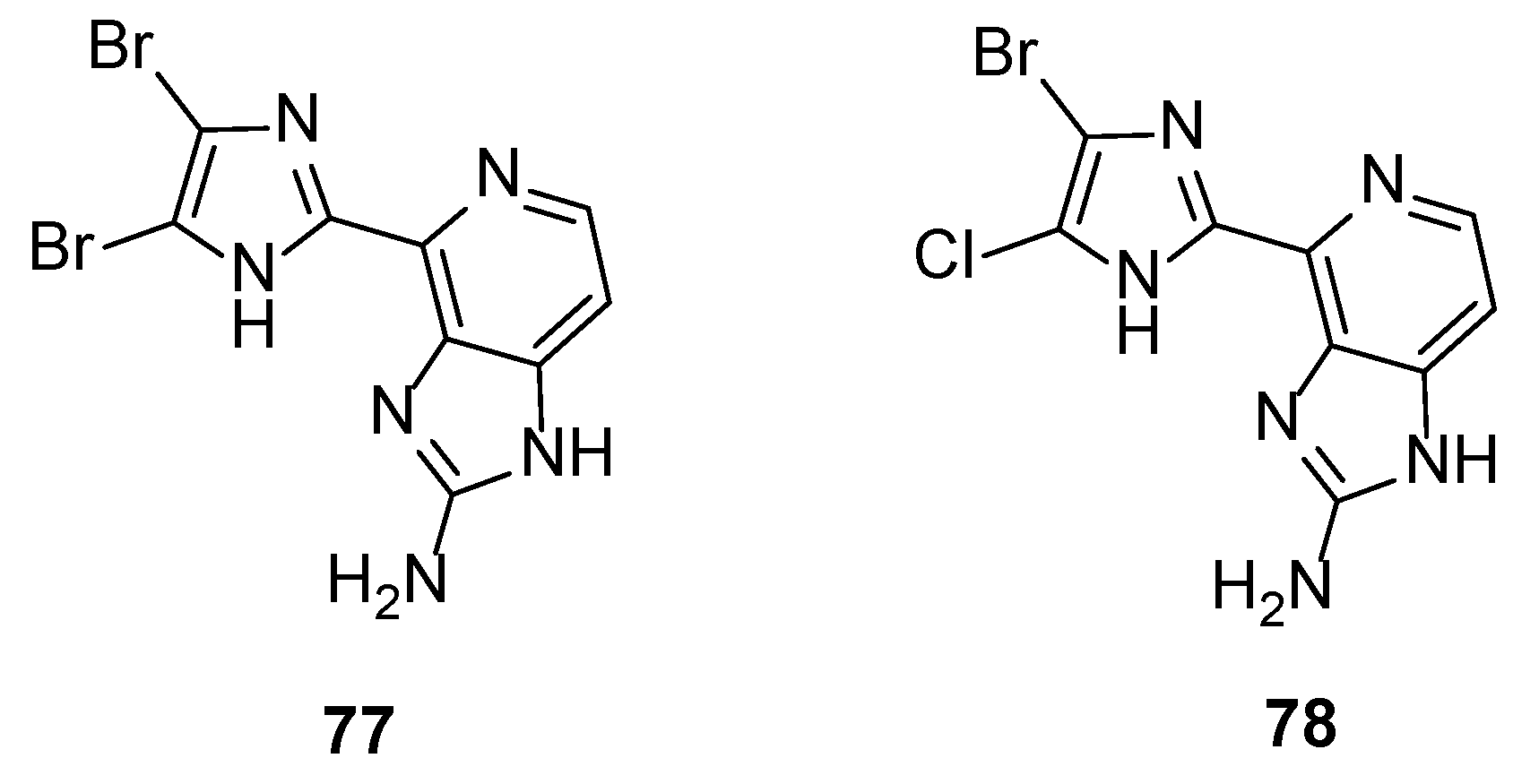
The biological outcomes of the aforementioned studies from Ando et al. produced a third generation of ten new analogues. The pyrrole ring was replaced by either imidazole, triazole, or tetrazole rings to further examine the role of the acidic proton of the pyrrole ring to the activity [41]. The results confirmed the previous findings that only analogues having a halogen at the C-2 position (Br or Cl) showed inhibitory activity. In particular, compounds 77 and 78 (Figure 6) were more potent than Ageladine A, with dibromoimidazole 77 being four-fold more active (IC50 = 0.86 μM). In addition, the presence of the -NH of the pyrrole is essential, since acidic triazole or tetrazole rings showed no or very weak activity. However, the presence of halogen atoms at the C-2 and the C-3 position seem to be more important to the inhibitory effect than the strength of the acidity of the -NH.
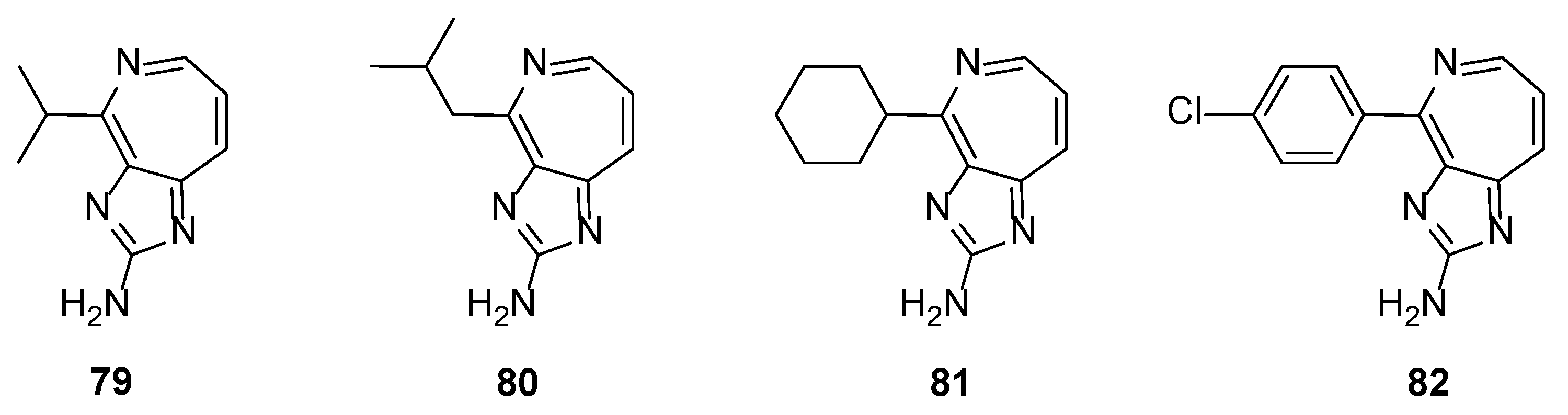
Further replacement of the pyrrole ring by other heterocycles (e.g., indole) or fused aromatic rings (e.g., naphthalene) or non-aromatic substituents (linear or cyclic) showed no inhibitory activity against MMPs. The same results were obtained even when the pyridine ring was replaced by an azepine ring, as can be exemplified in Figure 7 [42]. However, compounds 79–82 showed some interesting antiproliferative activity when tested against DU145 prostate, A2058 melanoma, and MDA-MB-435 breast cancer cells lines.
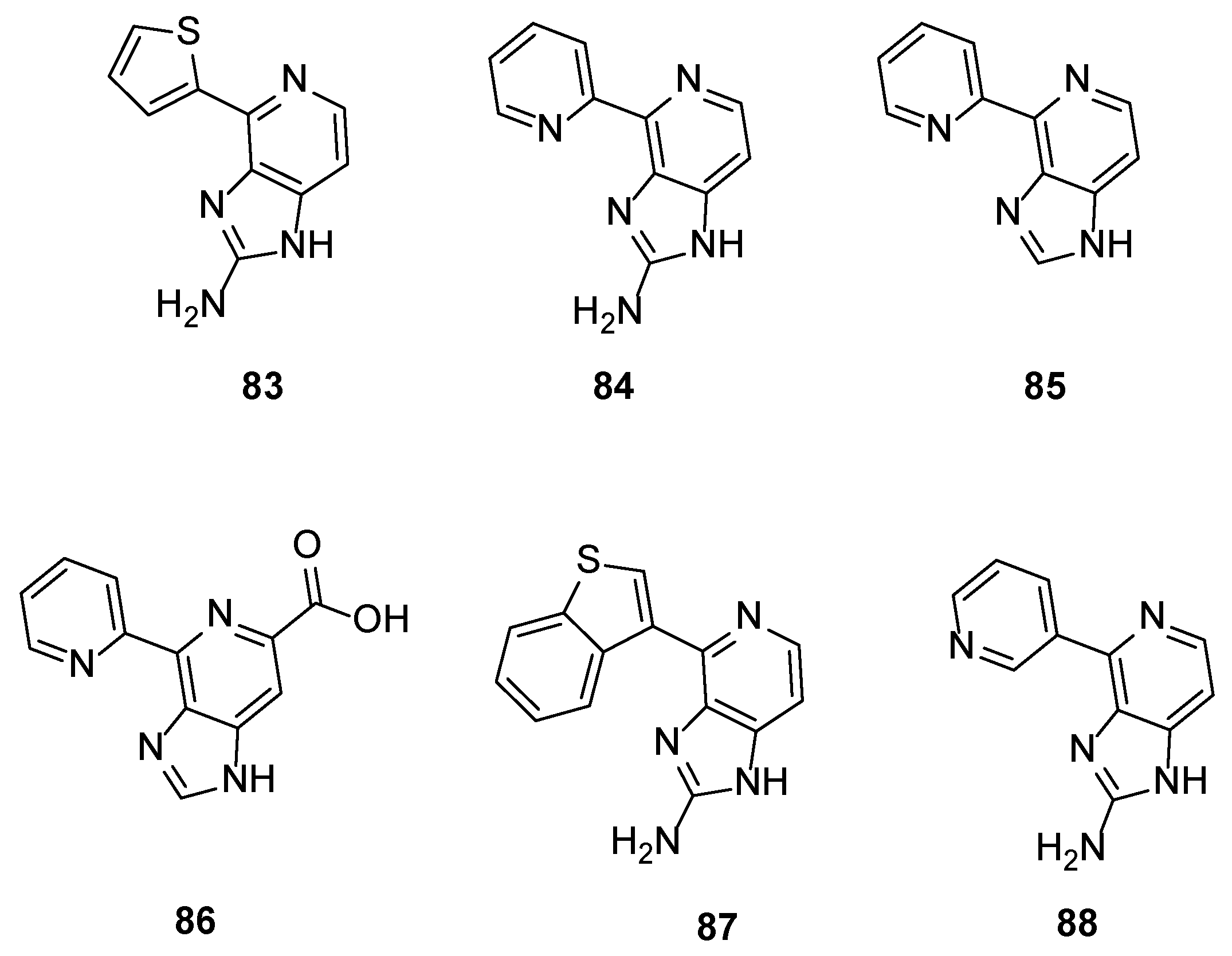
The application of the one-pot procedure developed by Karuso et al. [25] for the synthesis of Ageladine A led to a series of new analogues where the pyrrole ring was replaced by other heterocycles. These compounds were screened for their potential inhibitory activity against MMP-2 and MT1-MMP (MMP-14) and, as it was anticipated, with the exception of compound 83 (IC50 = 3.0 μg/mL for MMP-2 and 0.57 μg/mL for MT1-MMP), showed reduced activity compared to Ageladine A (IC50 = 1.7 μg/mL for MMP-2 and 0.2 μg/mL for MT1-MMP).
The natural product and some selected derivatives were also tested for their antiangiogenic activity. Surprisingly, 2-pyridine analogues 84, 85 and 86 (IC50 = 8, 19 and 21 μg/mL, respectively) and also benzothiophene analogue 87 (IC50 = 17 μg/mL) showed better antiangiogenic activity than Ageladine A (IC50 = 24 μg/mL). On the other hand, 3-pyridine derivative 88 showed no activity indicating the importance of the C-2 substitution of the pyridine core (Figure 8).
Ageladine A and compound 84 were further screened against 402 kinases. Both compounds showed similar profile by selectively inhibiting the same kinases, namely, DYRK1A, DYRK2, TYRK2 (JH2 domain), and YSK4. Collectively, it is reported for the first time, that the angiogenic activity of Ageladine A is not associated with MMP inhibition, but rather with kinase inhibitory activity. This remarkable conclusion renders Ageladine A as a very promising scaffold for the development of new potent and selective kinase inhibitors.
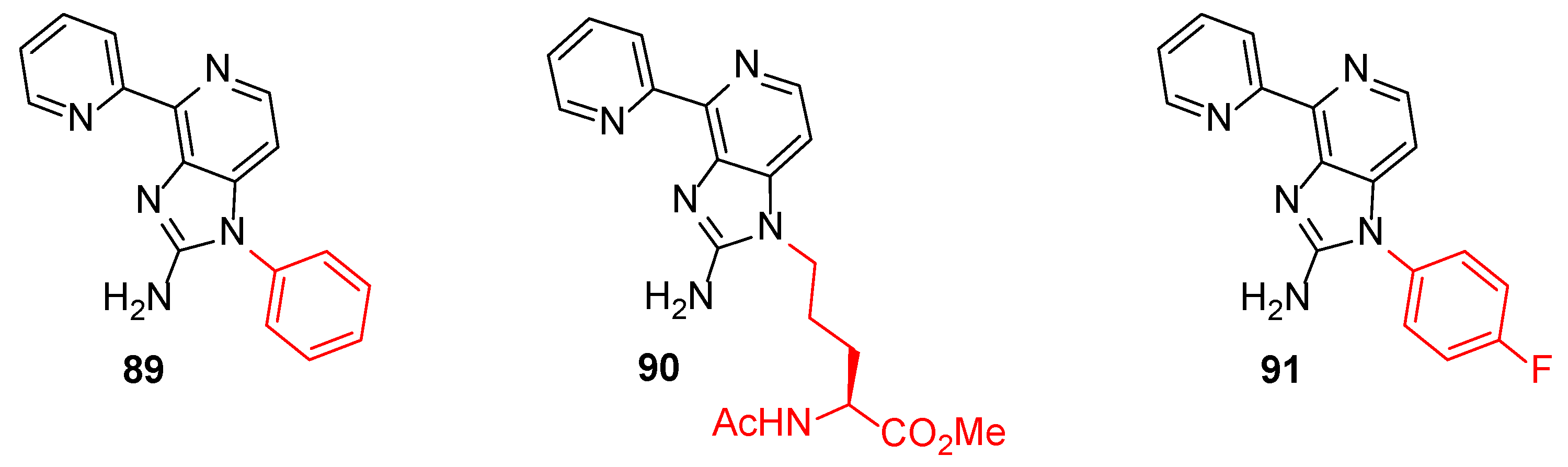
DYRK1A, a serine/threonine kinase, is associated with the suppression of the neural proliferation and differentiation and therefore is connected with pathogenesis, such as Alzheimer’s disease [43]. Prompted by the observation that Ageladine A and its analogue 84 inhibit this kinase, Tanaka et al. synthesized a series of 20 N1-substituted Ageladine A derivatives to study their potency to inhibit DYRK1A and to differentiate neural stem cells [30]. The results indicated that the substitution of Ageladine at the N1-position clearly eliminated the DYRK1A inhibitory activity of the compounds. At the same time, compounds 89 and 90 (Figure 9) were found to promote the neural differentiation and were more effective than harmine, which is a known DYRK1A inhibitor. On the other hand, compound 91 suppressed the differentiation of neurons while also showing negligible inhibitory activity of DYRK1A. These findings showed, beyond any doubt, that the differentiation–modulation activity of these compounds is DYRK1A-independent. It is very important to say that this modulation of neuron cells did not affect the astrocyte differentiation.
To this date, none of the synthesized analogs of Ageladine A showed better inhibitory activity against MMP-2. However, a number of compounds exhibited better inhibitory activity against MMP-12 compared to the natural metabolite. The following, Table 1, summarizes the in vitro activity of Ageladine A and its analogs against MMP-12.
Furthermore, a number of analogs exhibited better antiangiogenic activity compared to parent natural metabolite. The structures and the activity of these compounds are summarized in Table 2.
4. Conclusions
Marine sponges are an inexhaustible source of secondary metabolites offering both amazing structural diversity and a wide range of biological applications. Pyrrole alkaloids are one of the most prominent classes of such compounds. Ageladine A, derived by the marine sponge Agelas nakamurai, is a pyrrole-imidazole compound with very interesting biological activity as a MMP inhibitor and as a potent antiangiogenic compound. The elucidation of its structure and its biological profile motivated a number of research groups to describe efficient methods for the total synthesis of this natural product and to further expand their studies towards the synthesis of several derivatives in order to identify the structural elements responsible for such activity. Despite the fact that all total syntheses are inspired in terms of synthetic methodology, they suffer from low overall yields that render the synthesis of Ageladine A in gram-scale rather unattractive. Hence, there is still need for the discovery of new more efficient synthesis of Ageladine A, which will probably generate even more potent analogs in the future.
In the context of these synthetic approaches, a small number of compounds with better inhibitory activity compared to Ageladine A were discovered. Additionally, a new biological activity as potent neuron cells modulators without affecting the differentiation of the astrocytes was reported. Interestingly, Ageladine A has also the ability to stain fast-moving vesicles in live nerve cells, hence it can be used in fluorescence microscopy.
The present review attempted to highlight the synthetic efforts made towards the synthesis of Ageladine A and its derivatives and to shed lights on the structural features, which are indispensable for the activity. Hence, this work aspires to motivate more scientists in the field of marine natural products to discover new, more potent Ageladine A derivatives either as kinase inhibitors or towards a new target.
Funding
This research received no external funding.
Data Availability Statement
All data presented in this review were retrieved from the articles cited therein.
Conflicts of Interest
The author declares no conflict of interest.
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2022, 39, 1122–1171. [Google Scholar] [CrossRef]
- Chu, M.-J.; Li, M.; Ma, H.; Li, P.-L.; Li, G.-Q. Secondary metabolites from marine sponges of the genus Agelas: A comprehensive update insight on structural diversity and bioactivity. RSC Adv. 2022, 12, 7789–7820. [Google Scholar] [CrossRef] [PubMed]
- Manconi, R.; Perino, E.; Pronzato, R. A new species of Agelas from the Zanzibar Archipelago, western Indian Ocean (Porifera, Demospongiae). ZooKeys 2016, 553, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroif-Grégoire, C.; Appenzeller, J.; Debitus, C.; Zaparucha, A.; Al-Mourabit, A. Debromokeramadine from the Marine Sponge Agelas cf. mauritiana: Isolation and Short Regioselective and Flexible Synthesis. Tetrahedron 2015, 71, 3609–3613. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Y.; Gu, B.-B.; Yang, F.; Jiao, W.-H.; Hu, G.-H.; Yu, H.-B.; Han, B.-N.; Zhang, W.; Shen, Y.; et al. Antifungal Bromopyrrole Alkaloids from the South China Sea Sponge Agelas sp. Tetrahedron 2016, 72, 2964–2971. [Google Scholar] [CrossRef]
- Scala, F.; Fattorusso, E.; Menna, M.; Taglialatela-Scafati, O.; Tierney, M.; Kaiser, M.; Tasdemir, D. Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites. Mar. Drugs 2010, 8, 2162–2174. [Google Scholar] [CrossRef] [Green Version]
- Tempone, A.G.; Pieper, P.; Borborema, S.E.T.; Thevenard, F.; Lago, J.H.G.; Croft, S.L.; Anderson, E.A. Marine alkaloids as bioactive agents against protozoal neglected tropical diseases and malaria. Nat. Prod. Rep. 2021, 38, 2214–2235. [Google Scholar] [CrossRef]
- Fattorusso, E.; Taglialatela-Scafati, O. Two Novel Pyrrole-Imidazole Alkaloids from the Mediterranean Sponge Agelas oroides. Tetrahedron Lett. 2000, 41, 9917–9922. [Google Scholar] [CrossRef]
- Vik, A.; Hedner, E.; Charnock, C.; Samuelsen, Ø.; Larsson, R.; Gundersen, L.-L.; Bohlin, L. (+)-Agelasine D: Improved Synthesis and Evaluation of Antibacterial and Cytotoxic Activities. J. Nat. Prod. 2006, 69, 381–386. [Google Scholar] [CrossRef]
- Hertiani, T.; Edrada-Ebel, R.; Ortlepp, S.; van Soest, R.W.M.; de Voogd, N.J.; Wray, V.; Hentschel, U.; Kozytska, S.; Müller, W.E.G.; Proksch, P. From Anti-Fouling to Biofilm Inhibition: New Cytotoxic Secondary Metabolites from Two Indonesian Agelas Sponges. Bioorg. Med. Chem. 2010, 18, 1297–1311. [Google Scholar] [CrossRef]
- Zidar, N.; Žula, A.; Tomašič, T.; Rogers, M.; Kirby, R.W.; Tytgat, J.; Peigneur, S.; Kikelj, S.D.; Janez, I.; Mašič, L.P. Clathrodin, hymenidin and oroidin, and their synthetic analogues as inhibitors of the voltage-gated potassium channels. Eur. J. Med. Chem. 2017, 139, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Nakao, Y.; Matsunaga, S.; Seiki, M.; Itoh, Y.; Yamashita, J.; van Soest, R.W.M.; Fusetani, N. Ageladine A: An Antiangiogenic Matrixmetalloproteinase Inhibitor from the Marine Sponge Agelas nakamurai. J. Am. Chem. Soc. 2003, 125, 15700–15701. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.; Willoughby, R.; Shirley, A.; Pomponi, S.A.; Kerr, R.G. Biosynthetic Studies of the Alkaloid, Stevensine, in a Cell Culture of the Marine Sponge Teichaxinella morchella. Tetrahedron Lett. 1999, 40, 4775–4778. [Google Scholar] [CrossRef]
- Al-Mourabit, A.; Potier, P. Sponge’s Molecular Diversity Through the Ambivalent Reactivity of 2-Aminoimidazole: A Universal Chemical Pathway to the Oroidin-Based Pyrrole-Imidazole Alkaloids and Their Palau’amine Congeners. Eur. J. Org. Chem. 2001, 2001, 237–243. [Google Scholar] [CrossRef]
- Vergne, C.; Boury-Esnault, N.; Perez, T.; Martin, M.-T.; Adeline, M.-T.; Dau, E.T.H.; Al-Mourabit, A.; Verpacamides, A.-D. A Sequence of C11N5 Diketopiperazines Relating Cyclo(Pro-Pro) to Cyclo(Pro-Arg), from the Marine Sponge Axinella vaceleti: Possible Biogenetic Precursors of Pyrrole-2-aminoimidazole Alkaloids. Org. Lett. 2006, 8, 2421–2424. [Google Scholar] [CrossRef]
- Bickmeyer, U.; Grube, A.; Klings, K.W.; Köck, M.; Ageladine, A. A pyrrole-imidazole alkaloid from marine sponges, is a pH sensitive membrane permeable dye. Biochem. Biophys. Res. Commun. 2008, 373, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Bickmeyer, U.; Heine, M.; Podbielski, I.; Münda, D.; Köck, M.; Karuso, P. Tracking of fast moving neuronal vesicles with ageladine A. Biochem. Biophys. Res. Commun. 2010, 402, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meketa, M.L.; Weinreb, S.M. Total Synthesis of Ageladine A, an Angiogenesis Inhibitor from the Marine Sponge Agelas nakamurai. Org. Lett. 2006, 8, 1443–1446. [Google Scholar] [CrossRef]
- Vargas, D.F.; Larghi, E.L.; Kaufman, T.S. The 6p-azaelectrocyclization of azatrienes. Synthetic applications in natural products, bioactive heterocycles, and related fields. Nat. Prod. Rep. 2019, 36, 354–401. [Google Scholar] [CrossRef]
- Schumacher, R.W.; Davidson, B.S. Synthesis of didemnolines A-D, N9-substituted β-carboline alkaloids from the marine ascidian Didemnum sp. Tetrahedron 1999, 55, 935–942. [Google Scholar] [CrossRef]
- Iddon, B.; Khan, N. Azoles. Part 5. Metal-halogen exchange reactions of polybromoimidazoles. J. Chem. Soc. Perkin Trans. 1987, 1, 1445–1451. [Google Scholar] [CrossRef]
- Groziak, M.P.; Wei, L. Regioselective formation of imidazol-2-yllithium, imidazol-4-yllithium, and imidazol-5-yllithium species. J. Org. Chem. 1991, 56, 4296–4300. [Google Scholar] [CrossRef]
- Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. Catalysts for Suzuki−Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. [Google Scholar] [CrossRef] [PubMed]
- Shengule, S.R.; Karuso, P. Concise Total Synthesis of the Marine Natural Product Ageladine, A. Org. Lett. 2006, 8, 4083–4084. [Google Scholar] [CrossRef]
- Shengule, S.R.; Loa-Kum-Cheung, W.L.; Christopher, R.; Parish, C.R.; Blairvacq, M.; Meijer, L.; Nakao, Y.; Karuso, P. A One-Pot Synthesis and Biological Activity of Ageladine A and Analogues. J. Med. Chem. 2011, 54, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Ando, N.; Terashima, S. Synthesis and matrix metalloproteinase (MMP)-12 inhibitory activity of ageladine A and its analogs. Bioorg. Med. Chem. Lett. 2007, 17, 4495–4499. [Google Scholar] [CrossRef] [PubMed]
- Meketa, M.L.; Weinreb, S.M. A New Total Synthesis of the Zinc Matrix metalloproteinase Inhibitor Ageladine A Featuring a Biogenetically Patterned 6π-2-Azatriene Electrocyclization. Org. Lett. 2007, 9, 853–855. [Google Scholar] [CrossRef]
- Meketa, M.L.; Weinreb, S.M. A convergent total synthesis of the marine sponge alkaloid Ageladine A via a strategic 6p-2-azatriene electrocyclization. Tetrahedron 2007, 63, 9112–9119. [Google Scholar] [CrossRef]
- Jiang, L.; Job, G.E.; Klapars, A.; Buchwald, S.L. Copper-Catalyzed Coupling of Amides and Carbamates with Vinyl Halides. Org. Lett. 2003, 5, 3667–3669. [Google Scholar] [CrossRef]
- Iwata, T.; Otsuka, S.; Tsubokura, K.; Kurbangalieva, A.; Arai, D.; Fukase, K.; Nakao, Y.; Tanaka, K. One-Pot Evolution of Ageladine A through a Bio-Inspired Cascade towards Selective Modulators of Neuronal Differentiation. Chem. Eur. J. 2016, 22, 14707–14716. [Google Scholar] [CrossRef]
- Iwata, T.; Fukase, K.; Nakao, Y.; Tanaka, K. Efficient Synthesis of Marine Alkaloid Ageladine A and its Structural Modification for Exploring New Biological Activity. J. Synth. Org. Chem. Jpn. 2020, 78, 51–59. [Google Scholar] [CrossRef]
- Oe, T.; Lee, S.H.; Elipe, M.V.S.; Arison, B.H.; Blair, I.A. A Novel Lipid Hydroperoxide-Derived Modification to Arginine. Chem. Res. Toxicol. 2003, 16, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Tolle, C.; Fresia, M.; Lindel, T. Aza-BODIPY Route to Ageladine, A. Org. Lett. 2022, 24, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lou, Y.; Chen, Q.; Li, L.; Zhuang, X.; Li, Y. Synthesis of [1,2,5]thiadiazolo[3,4-c]pyridine: A Novel analogue to BT. Adv. Mater. Res. 2011, 335−336, 90–95. [Google Scholar] [CrossRef]
- Bender, T.; von Zezschwitz, P. Total Synthesis of 4-Acetyl-1,3-dihydroimidazo[4,5-c]pyridin-2-one, a New Microbial Metabolite from Streptomyces Species. Nat. Prod. Commun. 2009, 4, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and preparation of new palladium precatalysts for C-C and C-N cross-coupling reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Khalil, R.A. Matrix Metalloproteinase Inhibitors as Investigational and Therapeutic Tools in Unrestrained Tissue Remodeling and Pathological Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 148, 355–420. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Meketa, M.L.; Weinreb, S.M.; Nakao, Y.; Fusetani, N. Application of a 6π-1-Azatriene Electrocyclization Strategy to the Total Synthesis of the Marine Sponge Metabolite Ageladine A and Biological Evaluation of Synthetic Analogues. J. Org. Chem. 2007, 72, 4892–4899. [Google Scholar] [CrossRef]
- Ando, N.; Terashima, S. Synthesis of novel ageladine A analogs showing more potent matrix metalloproteinase (MMP)-12 inhibitory activity than the natural product. Bioorg. Med. Chem. Lett. 2009, 19, 5461–5463. [Google Scholar] [CrossRef]
- Ando, N.; Terashima, S. Synthesis and Matrix Metalloproteinase-12 Inhibitory Activity of Ageladine A Analogs. Chem. Pharm. Bull. 2011, 59, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Nam, S.; Jove, R.; Yakushijin, K.; Horne, D.A. Synthesis and anticancer activities of ageladine A and structural analogs. Bioorg. Med. Chem. Lett. 2010, 20, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, J.; Gong, C.-X.; Hwang, Y.-W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed biosynthetic pathway of Ageladine A (6) by Fusetani et al. [12].
Figure 1.
Proposed biosynthetic pathway of Ageladine A (6) by Fusetani et al. [12].

Scheme 1.
Total synthesis of Ageladine A by Weinreb et al. [18].
Scheme 1.
Total synthesis of Ageladine A by Weinreb et al. [18].


Scheme 3.
Total synthesis of Ageladine A by Ando et al. [26].
Scheme 3.
Total synthesis of Ageladine A by Ando et al. [26].

Scheme 4.
Synthesis of the pyrrole fragment 31.


Scheme 6.
Final steps towards the second-generation total synthesis of Ageladine A by Weinreb et al. [27,28].

Scheme 7.
Total synthesis of Ageladine A by Tanaka et al. [30].
Scheme 7.
Total synthesis of Ageladine A by Tanaka et al. [30].

Scheme 8.
Total synthesis of Ageladine A by Lindel et al. [33].
Scheme 8.
Total synthesis of Ageladine A by Lindel et al. [33].

Figure 2.
N-methylated derivatives of Ageladine A.

Figure 3.
Structures of compounds 15, 19 and 62–64.

Figure 4.
Structures of compounds synthesized by Ando et al. [26].
Figure 4.
Structures of compounds synthesized by Ando et al. [26].

Figure 5.
Structures of the most active compounds 74–76 against MMP-12.

Figure 6.
Structures of the most active imidazole derivatives 77 and 78.

Figure 7.
Structures of the imidazo[4,5-c]azepin-2-amine derivatives 79–82.

Figure 8.
Structures of compounds 83–88.

Figure 9.
Structures of modulators 89–91.


Table 1.
Structures and inhibitory activity against MMP-12 of Ageladine A and its more potent analogs.











{kind=link}
{kind=link}
{kind=link}
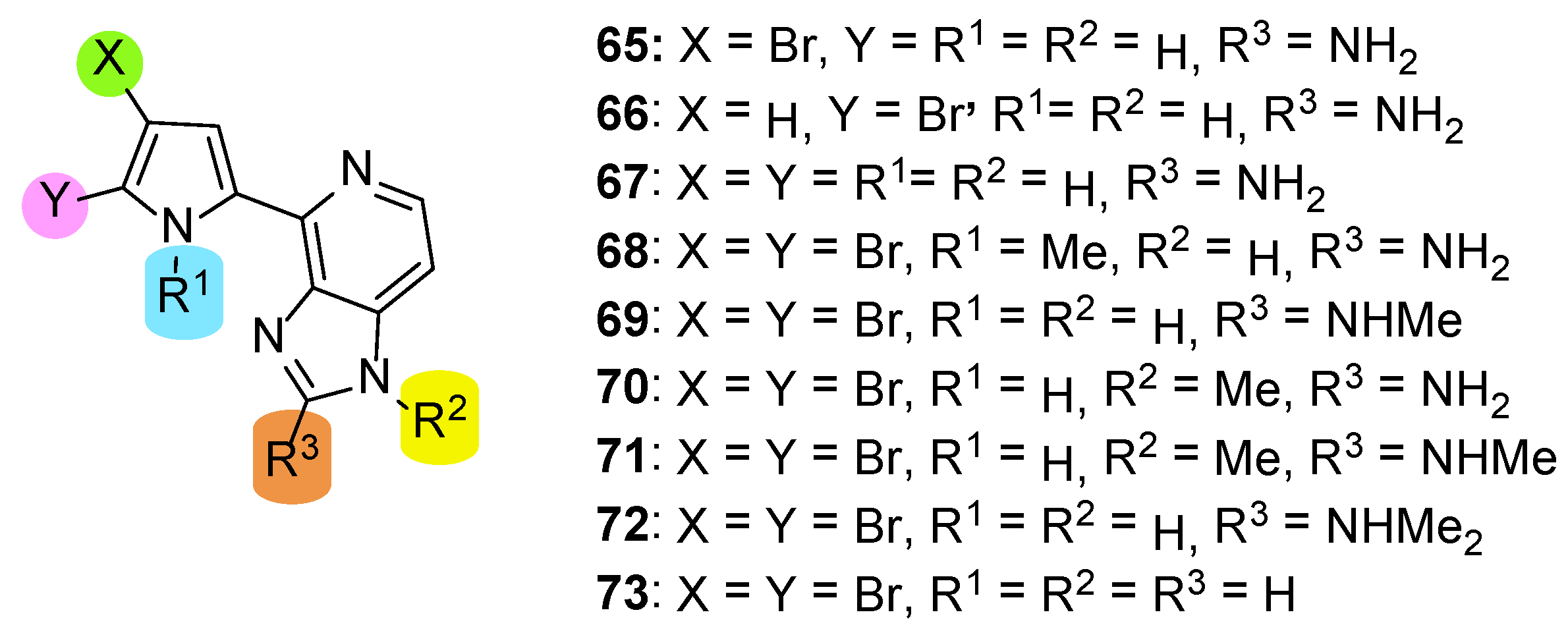{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Magoulas, G.E. Ageladine A, a Bromopyrrole Alkaloid from the Marine Sponge Agelas nakamurai. Compounds 2023, 3, 107-121. https://doi.org/10.3390/compounds3010010
AMA Style
Magoulas GE. Ageladine A, a Bromopyrrole Alkaloid from the Marine Sponge Agelas nakamurai. Compounds. 2023; 3(1):107-121. https://doi.org/10.3390/compounds3010010
Chicago/Turabian StyleMagoulas, George E. 2023. "Ageladine A, a Bromopyrrole Alkaloid from the Marine Sponge Agelas nakamurai" Compounds 3, no. 1: 107-121. https://doi.org/10.3390/compounds3010010