
Adsorption of Selected Molecules on (TiO2)20 Nano-Clusters: A Density-Functional-Theory Study


Abstract
:
1. Introduction
2. Theoretical Approach and Computational Details
2.1. Pure Functional PBE-SIESTA
2.2. Hybrid Functionals B3LYP and M06-L
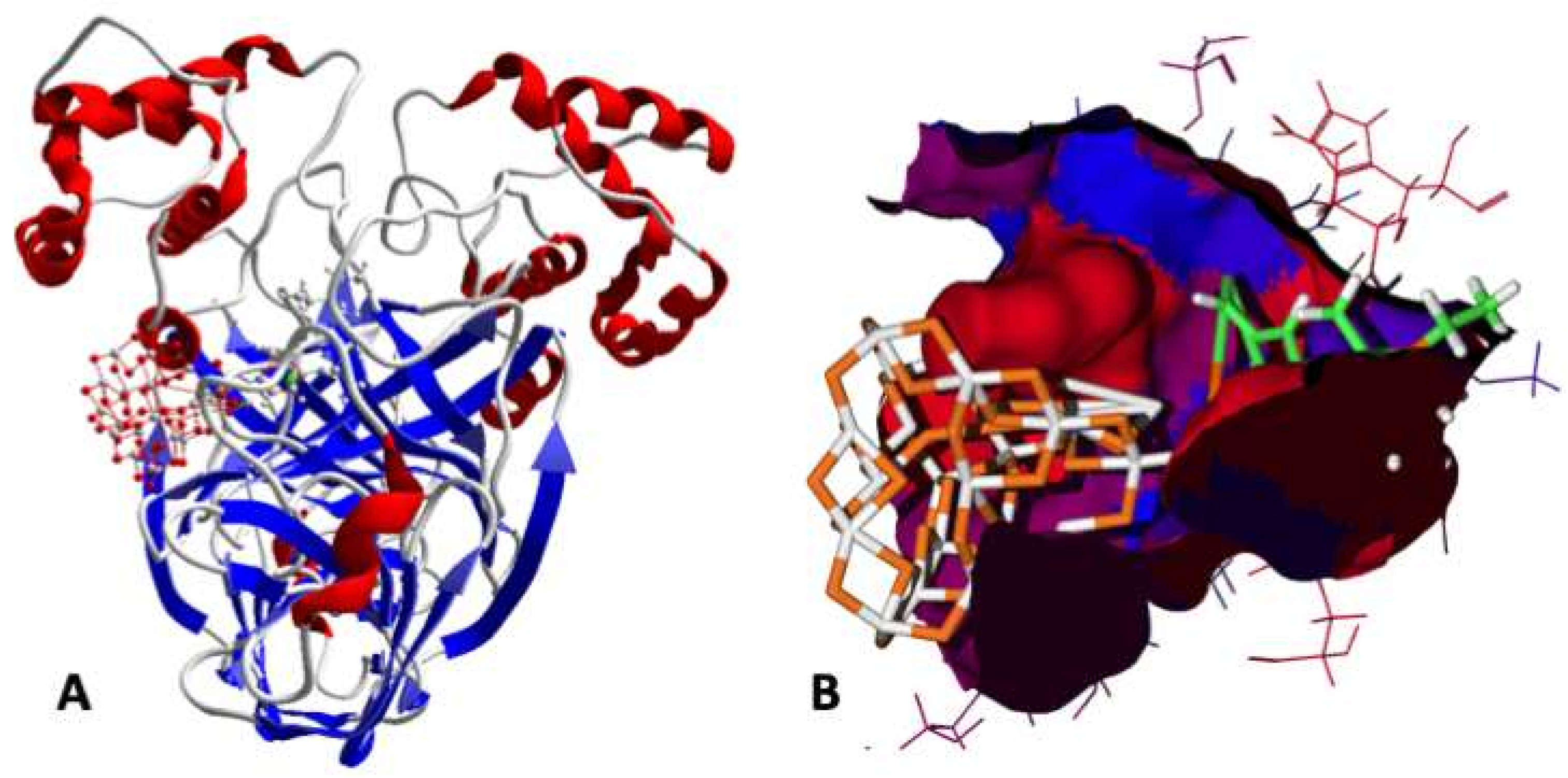
2.3. Activity against SARS-CoV-2 (Docking)
2.4. Chemical Energy Descriptors
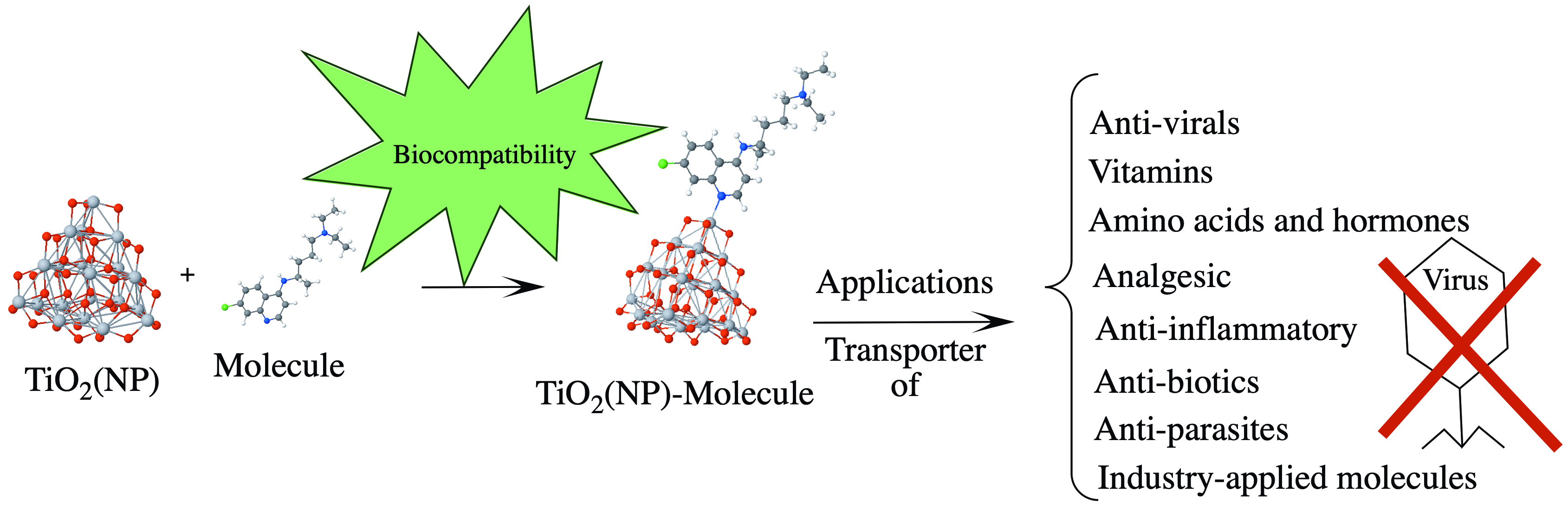
2.5. Condensed Fukuis Functions
3. Results
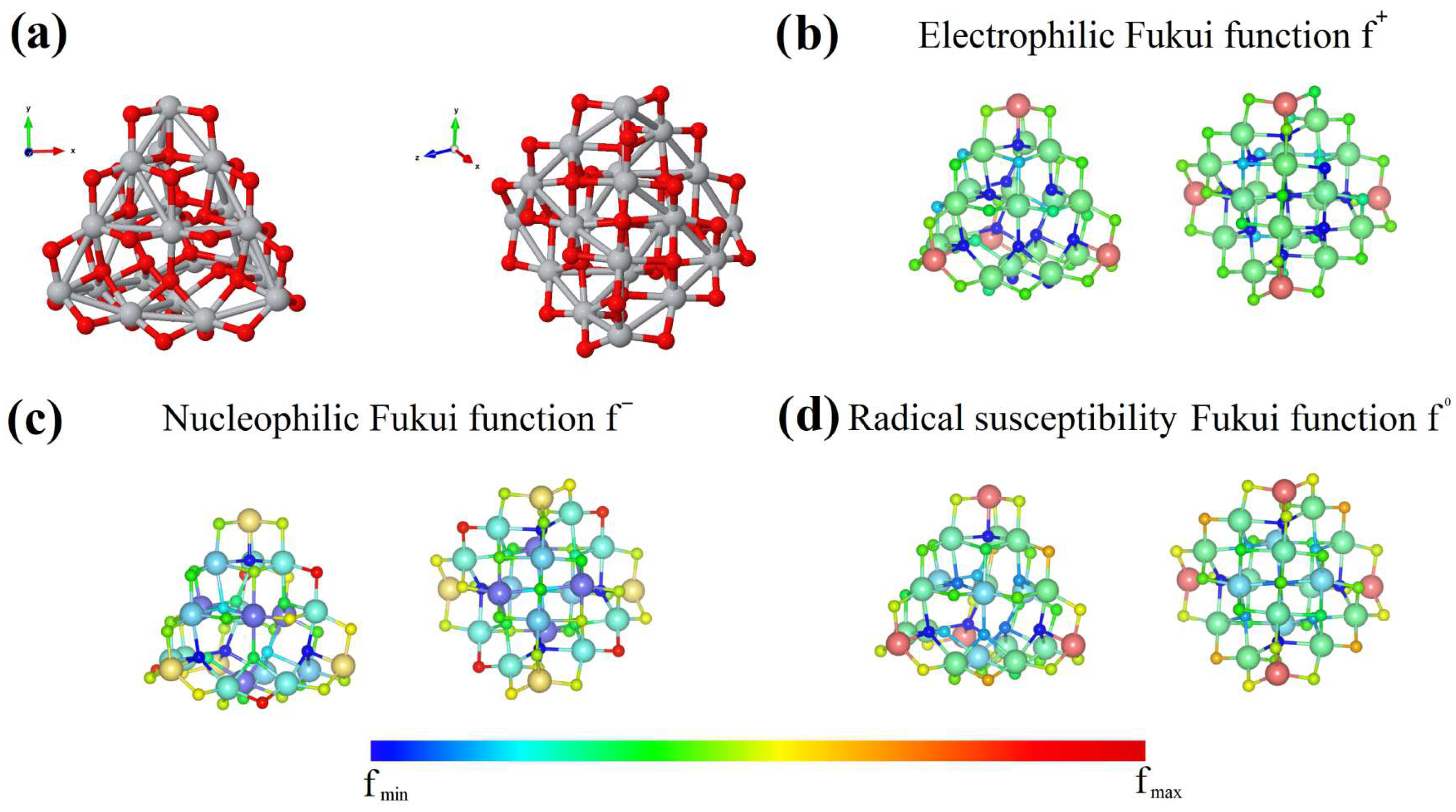
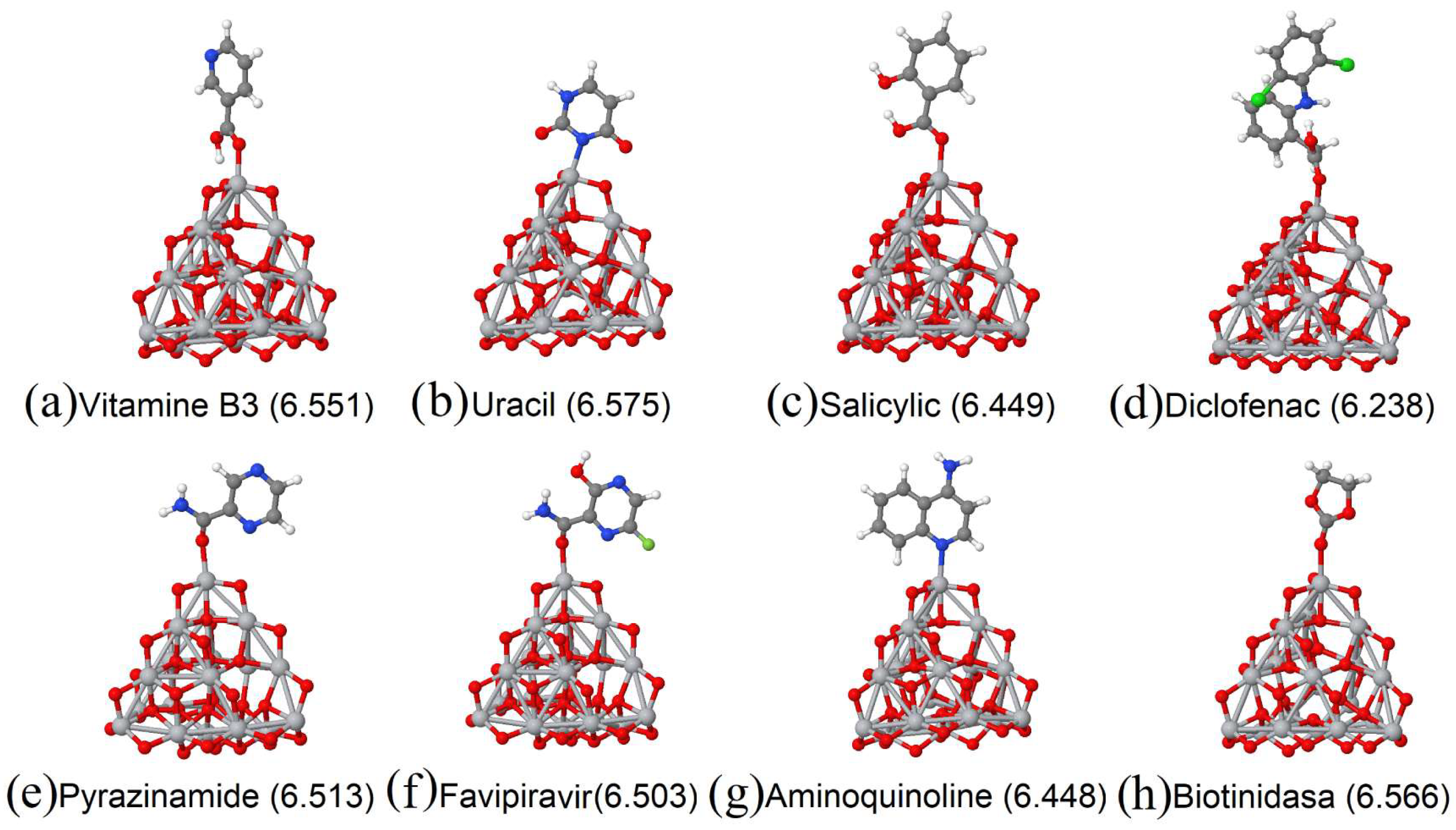
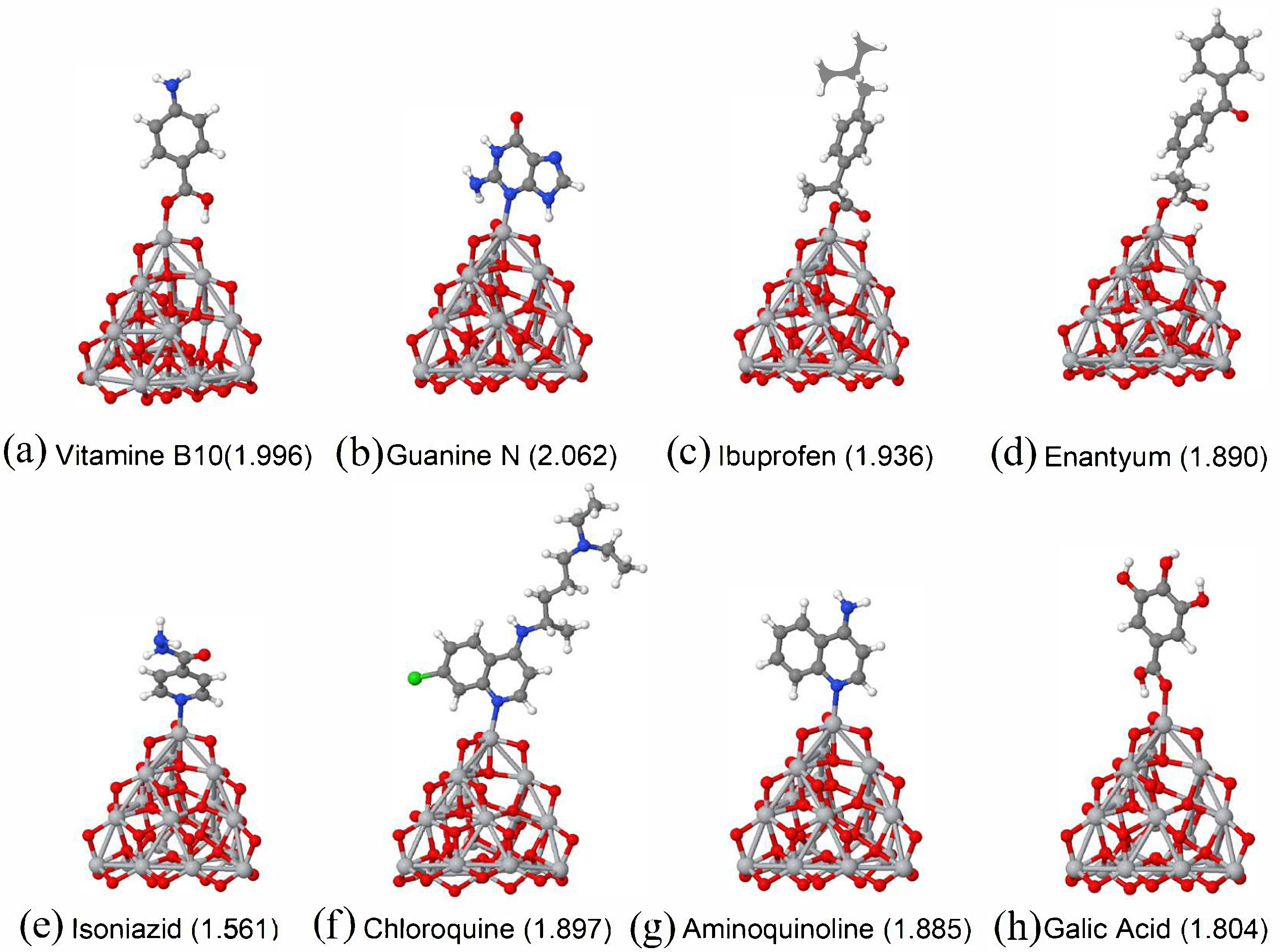
3.1. Vitamins
3.2. Amino Acids and/or Hormones
3.3. Analgesics and Anti-Inflammatory Drugs
3.4. Antibiotis
3.5. Industry and/or Food
3.6. Chemical Bond Size
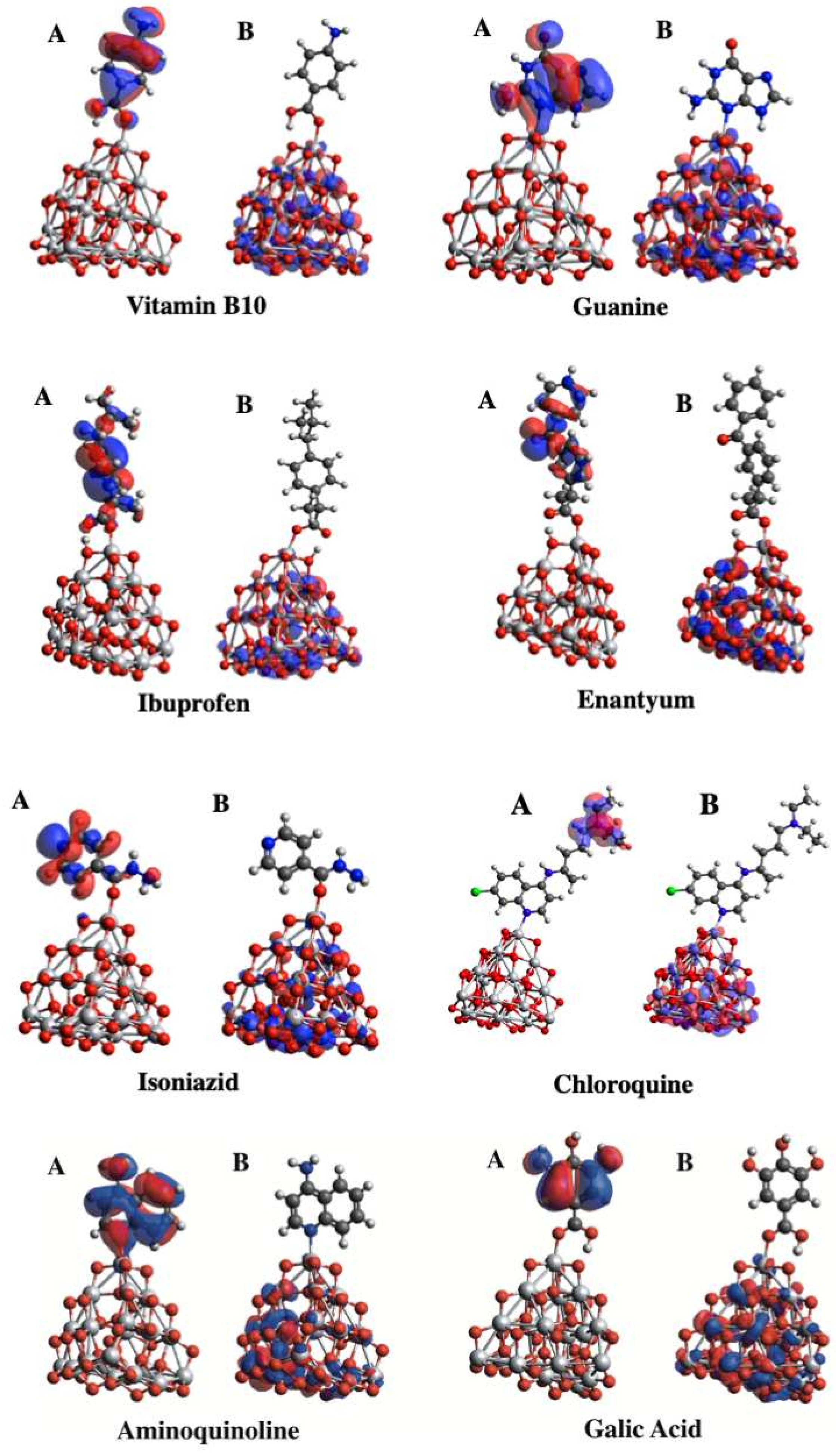
3.7. Molecular Orbitals Analysis
3.8. Comparison with Other Calculations
3.9. Activity against SARS-CoV-2 (Docking)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daimon, T.; Hirakawa, T.; Kitazawa, M.; Suetake, J.; Nosaka, Y. Formation of singlet molecular oxygen associated with the formation of superoxide radicals in aqueous suspensions of TiO2 photocatalysts. Appl. Catal. Gen. 2008, 340, 169–175. [Google Scholar] [CrossRef]
- Rajakumar, G.; Rahuman, A.; Roopan, S. Fungus-mediated biosynthesis and characteriza-tion of TiO2 nanoparticles and their activity against pathogenic bacteria, Spectrochim. Acta Part A 2012, 91, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.T.; Al-Khedhairy, A.A.; Musarrat, J. ZnO and TiO2 nanoparticles as novel antimicrobial agents for oral hygiene: A review. J. Nanopart. Res. 2015, 17, 276. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.; Reller, A. Photoinduced reactivity of titanium dioxide. Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Kamat, P.V. TiO2 Nanostructures: Recent Physical Chemistry Advances. J. Phys. Chem. C 2012, 116, 11849–11851. [Google Scholar] [CrossRef]
- Chen, M.; Straatsma, T.P.; Dixon, D.A. Molecular and Dissociative Adsorption of Water on (TiO2)n Clusters, n = 1–4. J. Phys. Chem. A 2015, 119, 11406–11421. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.; Shi, H.; Zhou, M.; Zhang, Y.; Chen, Q.; Zhang, Z. Preparation of elec-trospun Ag/TiO2 nanotubes with enhanced photocatalytic activity based on water/oil phase separation. Physica E 2017, 86, 103–110. [Google Scholar] [CrossRef]
- Piccinno, F.; Gottschalk, F.; Seeger, S.; Nowack, B. Industrial production quantities and uses of ten engineered nanomaterials in Europe and the world. J. Nanopart. Res. 2012, 14, 1109–1120. [Google Scholar] [CrossRef]
- Zhu, Z.; Cai, H.; Sun, D.-W. Titanium dioxide (TiO2) photocatalysis technology for nonthermal inactivation of microorganisms in foods. Trends Food Sci. Technol. 2018, 75, 23–35. [Google Scholar] [CrossRef]
- Rodríguez-González, V.; Obregón, S.; Patrón-Soberano, O.; Terashima, C.; Fujishima, A. An approach to the photocatalytic mechanism in the TiO2-nanomaterials microorganism interface for the control of infectious processes. Appl. Catal. B-Environ. 2020, 270, 118853. [Google Scholar] [CrossRef]
- Calle, I.D.L.; Menta, M.; Klein, M.; Séby, F. Screening of TiO2 and Au nanoparticles in cosmetics and determination of elemental impurities by multiple techniques (DLS, SP-ICP-MS, ICP-MS and ICP-OES). Talanta 2017, 171, 291–306. [Google Scholar] [CrossRef]
- Guo, M.-Z.; Chen, J.; Xia, M.; Wang, T.; Poon, C.S. Pathways of conversion of nitrogen oxides by nano TiO2 incorporated in cement-based materials. Build. Environ. 2018, 144, 412–418. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, J.-Y.; Li, H.-Q.; Zhao, A.Z.-J.; Wang, Y.; Zhang, K.-Q.; Sun, H.-T.; Lai, Y.-K. Recent advances on smart TiO2 nanotube platforms for sustainable drug delivery applications. Int. J. Nanomed. 2017, 12, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.; Subbiah, S.; Smith, E.; Kendall, R.J.; Ramkumar, S.S. Functional PVA/VB2/TiO2 Nanofiber Webs for Controlled Drug Delivery. ACS Appl. Bio Mater. 2019, 2, 5916–5929. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Granja, F.; Aguilera-del-Toro, R.H.; Vogel, E.E.; Cisternas, E. TiO2 nanoclusters adsorbed on surfaces: A density-functional-theoretic study. J. Phys. Chem. Solids 2021, 150, 109716. [Google Scholar] [CrossRef]
- Aguilera-del-Toro, R.H.; Aguilera-Granja, F.; Vogel, E.E. Structural and electronic properties of (TiO2)N nanowires: A density functional theory investigation. J. Phys. Chem. Solids 2018, 119, 175–182. [Google Scholar] [CrossRef]
- Salazar-Villanueva, M.; Bautista-Hernandez, A.; Chigo-Anota, E.; Valdez, S.; Vázquez-Cuchillo, O. Electronic and structural properties of Ti9XO20 (X = Ti, C, Si, Ge, Sn and Pb) clusters: A DFT study. Physica E 2015, 65, 120–124. [Google Scholar] [CrossRef]
- Ko, K.C.; Bromley, S.T.; Lee, J.Y.; Illas, F. Size-Dependent Level Alignment between Rutile and Anatase TiO2 Nanoparticles: Implications for Photocatalysis. J. Phys. Chem. Lett. 2017, 8, 5593–5598. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Shen, L.Y.; Hu, Y.H. Preparation of TiO2-Graphene Composite by a Two-Step Solvothermal Method and its Adsorption-Photocatalysis Property. Water Air. Soil Pollut. 2016, 227, 1–12. [Google Scholar] [CrossRef]
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Shi, M.; Shen, J.; Ma, H.; Li, Z.; Lu, X.; Li, N.; Ye, M. Preparation of graphene-TiO2 composite by hydrothermal method from peroxotitanium acid and its photocatalytic properties. Colloids Surf. A Phisicochem. Eng. Asp. 2012, 405, 30–37. [Google Scholar] [CrossRef]
- Wan, J.K.; Hyun-Jung, K. Titanium dioxide-graphene oxide composites with different ratios supported by Pyrex tube for photocatalysis of toxic aromatic vapors. Powder Technol. 2013, 250, 115–121. [Google Scholar]
- Martínez-Sánchez, C.; Montiel-González, F.; Díaz-Cervantes, E.; Rodríguez-González, V. Unraveling the strength interaction in a TiO2-Graphene photocatalytic nanocomposite synthesized by the microwave hydrothermal method. Mater. Sci. Semicon. Proc. 2019, 101, 262–271. [Google Scholar] [CrossRef]
- Umebayashi, T.; Yamaki, T.; Itoh, H.; Asai, K. Analysis of electronic structures of 3d transition metal-doped TiO2 based on band calculations. J. Phys. Chem. Solids 2002, 63, 1909–1920. [Google Scholar] [CrossRef]
- Hou, X.; Huang, M.; Wu, X.; Liu, A. First-principles calculations on implanted TiO2 by 3d transition metal ions. Sci. China Ser. G Phys. Mech. Astron. 2009, 52, 838–842. [Google Scholar] [CrossRef]
- Salazar-Villanueva, M.; Cruz-López, A.; Zaldívar-Cadena, A.A.; Tovar-Corona, A.; Guevara-Romero, M.L.; Vazquez-Cuchillo, O. Effect of the electronic state of Ti on M-doped TiO2 nanoparticles (M = Zn, Ga or Ge) with high photocatalytic activities: A experimental and DFT molecular study. Mater. Sci. Semicond. Process. 2017, 58, 8–14. [Google Scholar] [CrossRef]
- Rodríguez-Torres, C.E.; Cabrera, A.F.; Errico, L.A.; Duhalde, S.; Rentería, M.; Golmar, F.; Sánchez, F.H. XAS study of the local environment of impurities in doped TiO2 thin films. Physica B 2007, 398, 219–222. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Li, J.; Li, L.; Lin, S. First-principles study on transition metal-doped anatase TiO2. Nanoscale Res. Lett. 2014, 9, 46. [Google Scholar] [CrossRef]
- Aguilera-del-Toro, R.H.; Aguilera-Granja, F.; Vogel, E.E. Structural and electronic properties of (TiO2)10 clusters with impurities: A density functional theory investigation. J. Phys. Chem. Solid 2019, 135, 109107. [Google Scholar] [CrossRef]
- Lamiel-Garcia, O.; Cuko, A.; Calatayud, M.; Illas, F.; Bromley, S.T. Predicting size-dependent emergence of crystallinity in nanomaterials: Titania nanoclusters versus nanocrystals. Nanoscale 2017, 9, 1049. [Google Scholar] [CrossRef]
- U.S. Food & Drug. Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine and Hydroxychloroquine|FDA; Update FDA; FDA: Washington, DC, USA, 2020; pp. 1–2.
- Rolain, J.-M.; Colson, P.; Raoult, D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agent 2007, 30, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Salameh, P.; Hajj, A.; Badro, D.A.; Acouselwan, C.; Aoun, R.; Sacre, H. Mental Health Outcomes of the COVID-19 Pandemic and a Collapsing Economy: Perspectives from a Developing Country. Psichiatry Res. 2020, 294, 113520. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejon, P.; Sánchez-Portal, D.S. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Kleinman, L.; Bilander, D.M. Efficacious Form for Model Pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425–1428. [Google Scholar] [CrossRef]
- Aguilera-Granja, F.; Piotrowski, M.J.; Da Silva, J.L.F. Structural and electronic properties of TM23-pAgp (TM = Ni, Pd, and Pt) clusters in the dilute limit (p = 0–4): A density functional theory investigation. Eur. J. Phys. D 2013, 67, 33. [Google Scholar] [CrossRef]
- Aguilera-Granja, R.; Longo, R.C.; Gallego, L.J.; Vega, A. Structural and magnetic properties of X12Y (X, Y = Fe, Co, Ni, Ru, Rh, Pd, and Pt) nanoalloys. J. Chem. Phys. 2010, 132, 184507. [Google Scholar] [CrossRef]
- Alonso-Lanza, T.; Ayuela, A.; Aguilera-Granja, F. Substitutional 4d and 5d impurities in graphene. Phys. Chem. Chem. Phys. 2016, 18, 21913–21920. [Google Scholar] [CrossRef]
- Aguilera-Granja, F.; García-Fuente, A.; Vega, A. Comparative ab initio study of the structural, electronic, and magnetic trends of isoelectronic late 3d and 4d transition metal clusters. Phys. Rev. B 2008, 78, 134425. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in Fortran, 2nd ed.; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-Consistent Molecular Orbital Methods. 21. Small Split-Valence Basis Sets for First-Row Elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of Polarization and Diffuse Functions to the LANL2DZ Basis Set for P-Block Elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef]
- Mpiana, P.T.; Tshibangu, D.S.; Kilembe, J.T.; Gbolo, B.Z.; Mwanangombo, D.T.; Inkoto, C.L.; Lengbiye, E.M.; Mbadiko, C.M.; Matondo, A.; Bongo, G.N.; et al. Identification of potential inhibitors of SARS-CoV-2 main protease from Aloe vera compounds: A molecular docking study. Chem. Phys. Lett. 2020, 754, 137751. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, B.; Jin, Z.; Yang, H.; Rao, Z. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3, 2020th ed.; Protein Data Bank. 2020. Available online: https://www.rcsb.org/structure/6lu7 (accessed on 28 June 2022).
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Chou, K.-C.; Wei, D.-Q.; Zhong, W.-Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003, 308, 148–151. [Google Scholar] [CrossRef]
- Cortés-García, C.J.; Chacón-García, L.; Mejía-Benavides, J.E.; Díaz-Cervantes, E. Tackling the SARS-CoV-2 main protease using hybrid derivatives of 1,5-disubstituted tetrazole-1,2,3-triazoles: An in silico assay. J. Phys. Chem. 2020, 2, e10. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, P.; Pérez, P.; Contreras, R. On the condensed Fukui function. J. Chem. Phys. 2000, 113, 2544–2551. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, softness and the Fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Aguilera-Granja, F.; Vega, A.; Balbas, L.C. New structural and electronic properties of (TiO2)10. J. Chem. Phys. C 2016, 144, 234312. [Google Scholar] [CrossRef]
- Chul-Ko, K.; Lamiel-García, O.; Yong-Lee, J.; Illas, F. Performance of a modified hybrid functional in the simultaneous description of stoichiometric and reduced TiO2 polymorphs. Phys. Chem. Chem. Phys. 2016, 18, 12357–12367. [Google Scholar]
- Mignon, P.; Ugliengo, P.; Sodupe, M. Theoretical Study of the Adsorption of RNA/DNA Bases on the External Surfaces of Na+-Montmorillonite. J. Phys. Chem. C 2009, 113, 13741–13749. [Google Scholar] [CrossRef]
- Irrera, S.; de Leeuw, N.H. A density functional theory study of the adsorption of uracil on the Au(100) surface. Proc. R. Soc. A 2011, 467, 1959–1969. [Google Scholar] [CrossRef]
- Young, Y.S.; Kim, D.H.; Lee, H.J.; Kim, S. Dissociative adsorption of guanine on Ge(100). Chem. Commun. 2015, 51, 12815. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Johnson, E.R. Theoretical investigation of amino-acid adsorption on hydroxylated quartz surfaces: Dispersion can determine enantioselectivity. Phys. Chem. Chem. Phys. 2020, 22, 16571. [Google Scholar] [CrossRef] [PubMed]
- Urdaneta, I.; Keller, A.; Atabek, O.; Palma, J.L.; Finkelstein-Shapiro, D.; Tarakeshwar, P.; Mujica, V.; Calatayud, M. Direct Evidence of Chelated Geometry of Catechol on TiO2 by a Combined Solid State NMR and DFT Study. J. Phys. Chem. C 2014, 118, 20688–20693. [Google Scholar] [CrossRef] [Green Version]
- Balducci, G. The adsorption of glucose at the surface of anatase: A computational study. Chem. Phys. Lett. 2010, 494, 54–59. [Google Scholar] [CrossRef]
- Alver, Ö.; Parlak, C.; Umar, Y.; Ramasami, P. DFT/QTAIM analysis of favipiravir adsorption on pristine and silicon doped C20 fullerenes. Main Group Mater. Chem. 2019, 42, 143–149. [Google Scholar] [CrossRef]
- Rad, A.S.; Ardjmand, M.; Esfahani, M.R.; Khodashenas, B. DFT calculations towards the geometry optimization, electronic structure, infrared spectroscopy and UV-vis analyses of Favipiravir adsorption on the first-row transition metals doped fullerenes; a new strategy for COVID-19 therapy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 247, 119082. [Google Scholar] [CrossRef] [PubMed]
- Saikia, N.; Deka, R.C. Adsorption of isoniazid and pyrazinamide drug molecules onto nitrogen-doped single-wall carbon nanotubes: An ab initio study. Struct. Chem. 2014, 25, 593–605. [Google Scholar] [CrossRef]
- Saikia, N.; Pati, S.K.; Deka, R.C. First principles calculation on the structure and electronic properties of BNNTs functionalized with isoniazid drug molecule. Appl. Nanosci. 2012, 2, 389–400. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
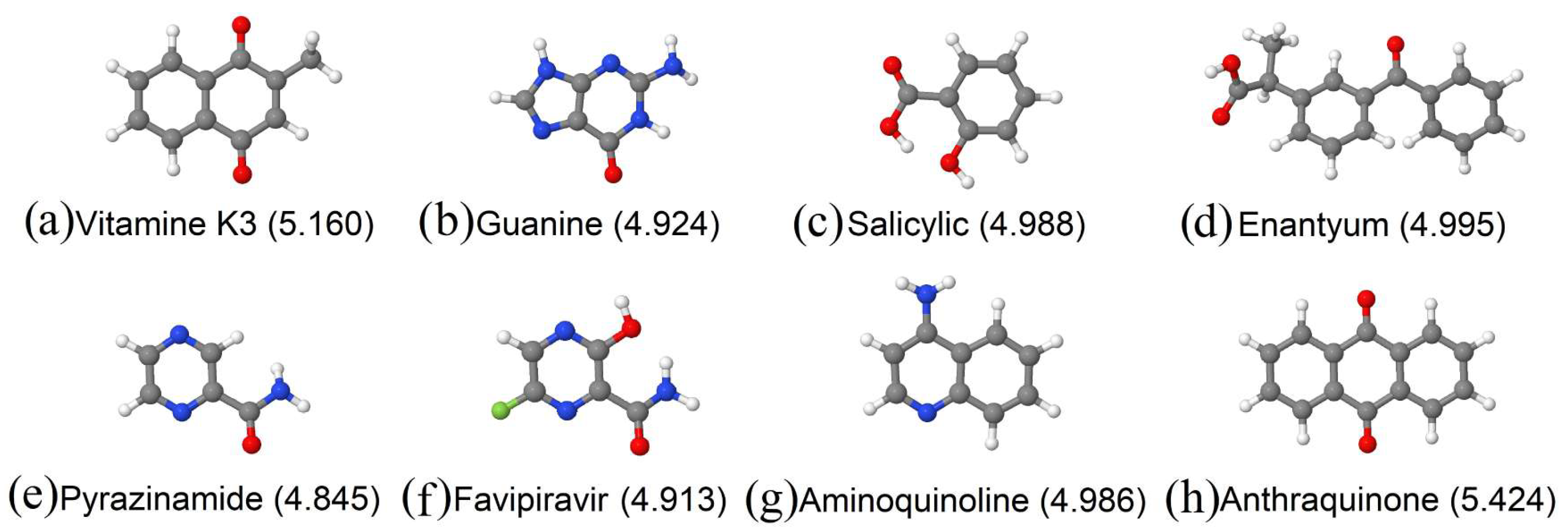{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule/Weight | E (eV/atom) Molecule | E (eV/atom) Complex | Ads. Ene. (eV) Molecule |
|---|---|---|---|
| Vitamin-like | |||
| B3(1) CHNO/122.125 | 4.862 (4.979) | 6.499 | 1.749 |
| B3(2) CHNO/123.109 | 5.014 (5.144) | 6.551 | 1.825 |
| B10 CHNO/137.136 | 4.875 (4.992) | 6.463 | 1.996 |
| B13 CHNO/156.096 | (5.191) | 6.542 | 1.801 |
| B6(1) CHNO/167.164 | 4.718 (4.781) | 6.335 | 1.315 |
| B6(2) CHNO/168.196 | 4.474 (4.529) | 6.208 | 1.324 |
| K3 CHO/172.180 | (5.225) | 6.450 | 1.362 |
| C CHO/176.124 | 4.573 (4.635) | 6.318 | 1.242 |
| Molecule | E (eV) Complex | Gap(H-L) (eV) Complex | Electric Dipole (Dbys) Molecule/Complex |
|---|---|---|---|
| Vitaminic-like | |||
| B3(1) CHNO | −5.842 | (0.882) | (, 10.306) |
| B3(2) CHNO | −5.461 | 2.634 (0.778) | (0.765, 4.697) |
| B10 CHNO | −5.089 | 2.133 (0.634) | (3.935, 1.681) |
| B13 CHNO | −6.061 | 1.911 (0.715) | (3.834, 0.840) |
| B6(1) CHNO | −6.246 | 2.129 (0.798) | (2.253, 11.052) |
| B6(2) CHNO | −5.549 | 2.129 (0.548) | (2.435, 10.731) |
| K3 CHO | −6.596 | 1.700 (0.878) | (1.042, 12.895) |
| C CHO | −5.895 | (0.828) | (4.096, 16.337) |
| Molecule/Weight | E (eV/atom) Molecule | E (eV/atom) Complex | Ads. Ene. (eV) Molecule |
|---|---|---|---|
| Amino acid and/or Hormones | |||
| DMG CHNO/103.120 | 4.132 (4.250) | 6.326 | 1.884 |
| Cytosine CHNO/111.100 | 4.708 (4.835) | 6.515 | 1.653 |
| Histamine CHN/111.150 | 4.318 (4.434) | 6.340 | 1.976 |
| Uracil CHNO/112.087 Bind O | 4.905 (5.049) | 6.574 | 1.735 |
| Bind N | (5.052) | 6.575 | 1.768 |
| Thymine CHNO/126.113 | 4.765 (4.862) | 6.475 | 1.456 |
| Adenine CHN/135.130 | 4.917 (5.053) | 6.514 | 2.037 |
| Glutamic acid CHNO/147.129 | 4.415 (4.479) | 6.302 | 1.230 |
| Guanine CHNO/151.126 Bind O | 4.924 (5.021) | 6.488 | 1.549 |
| Bind N | (5.053) | 6.495 | 2.062 |
| Dopamine CHNO/153.178 | 4.528 (4.580) | 6.262 | 1.150 |
| Histidine CHNO/155.157 | 4.525 (4.623) | 6.315 | 1.953 |
| Phenylalamine CHNO/165.189 | 4.663 (4.737) | 6.285 | 1.708 |
| Molecule | E (eV) Complex | Gap(H-L) (eV) Complex | Electric Dipole (Dbys) Molecule/Complex |
|---|---|---|---|
| Amino acid and/or Hormones | |||
| DMG CHNO | −5.252 | 1.492 (0.397) | (1.824, 5.005) |
| Cytosine CHNO | −5.562 | 3.068 (0.882) | (, ) |
| Histamine CHN | −5.093 | 2.496 (0.511) | (2.662, 13.156) |
| UracilCHNO Bind O | −6.236 | (0.863) | (4.295, 11.535) |
| Bind N | −6.644 | 2.908 (0.788) | (4.295, 8.581) |
| Thymine CHNO | −5.811 | (0.886) | (4.316, 13.366) |
| Adenine CHN | −6.265 | 2.467 (0.656) | (2.611, 7.411) |
| Glutamic acid CHNO | −5.750 | 2.325 (0.559) | (2.420, 10.884) |
| Guanine CHNO Bind O | −5.670 | (0.863) | (, ) |
| Bind N | −5.493 | 1.749 (0.472) | (, 3.097) |
| Dopamine CHNO | −5.315 | 1.586 (0.394) | (2.275, 6.786) |
| Histidine CHNO | −5.531 | 1.787 (0.426) | (5.384, 10.078) |
| Phenylalamine CHNO | −5.926 | 1.973 (0.478) | (1.901, 5.031) |
| Molecule/Weight | E (eV/atom) Molecule | E (eV/atom) Complex | Ads. Ene. (eV) Molecule |
|---|---|---|---|
| Analgesics | |||
| Salicylic CHO/138.120 | (5.076) | 1.400 | |
| Paracetamol CHNO/151.163 | 4.744 (4.827) | 6.366 | 1.651 |
| Aspirin CHO/180.157 | 4.966 (5.033) | 6.401 | 1.407 |
| Ibuprofen CHO/206.281 | 4.536 (4.595) | 6.068 | 1.936 |
| Tramadol CHNO/263.375 | 4.391 (4.427) | 5.841 | 1.575 |
| Anti-inflammatory | |||
| Enantyum CHO/254.285 | (5.052) | 6.231 | 1.890 |
| Diclofenac CHNOCl/296.147 | 4.897 (4.957) | 1.811 |
| Molecule | E (eV) Complex | Gap(H-L) (eV) Complex | Electric Dipole (Dbys) Molecule/Complex |
|---|---|---|---|
| Analgesics | |||
| Salicylic CHO | −6.547 | (0.799) | (, 5.092) |
| Paracetamol CHNO | −5.416 | 2.569 (0.729) | (3.362, ) |
| Aspirin CHO | −6.517 | (0.806) | (4.570, 5.545) |
| Ibuprofen CHO | −5.437 | 1.713 (0.408) | (1.708, 1.839) |
| Tramadol CHNO | −4.865 | 0.897 (0.268) | (2.136, 9.487) |
| Anti-inflammatory drugs | |||
| Enantyum CHO | -5.3355 | 1.464 (0.517) | (1.759, 3.084) |
| Diclofenac CHNOCl | −6.1151 | (0.821) | (, ) |
| Molecule/Weight | E (eV/atom) Molecule | E (eV/atom) Complex | Ads. Ene. (eV) Molecule |
|---|---|---|---|
| Antibiotics | |||
| Pyrazinamide CHNO/123.113 | 4.845 (4.944) | 6.513 | 1.379 |
| Isoniazid CHNO/137.139 Bind O | 4.698 (4.788) | 6.418 | 1.544 |
| Bind N | (4.789) | 6.418 | 1.561 |
| Anti-virals | |||
| Favipiravir CHNOF/157.103 | 4.913 (5.000) | 6.503 | 1.298 |
| Chloroquine (CH.) CHNCl/319.872 | 4.440 (4.480) | 5.813 | 1.897 |
| Hydroxy CH. CHNOCl/335.876 | 4.534 (4.572) | 5.854 | 1.856 |
| Anti-parasites | |||
| Aminoquinoline CHN/144.173 | 4.986 (5.085) | 6.448 | 1.885 |
| Metrodinazole CHNO/171.156 | 4.458 (4.510) | 6.265 | 1.097 |
| Amodiaquinine CHNOCl/355.866 | 4.705 (4.735) | 5.937 | 1.396 |
| Molecule | E (eV) Complex | Gap(H-L) (eV) Complex | Electric Dipole (Dbys) Molecule/Complex |
|---|---|---|---|
| Antibiotics | |||
| Pyrazinamide CHNO | −5.933 | 2.678 (0.923) | (, ) |
| Isoniazid CHNO Bind O | −5.653 | (0.822) | (2.238, 11.826) |
| Bind N | −5.8097 | 2.557 (0.755) | (2.238, 9.994) |
| Anti-virals | |||
| Favipiravir CHNOF | −5.7693 | (0.883) | (5.432, 17.269) |
| Chloroquine (CH.) CHNCl | −4.8366 | 1.427 (0.510) | (, ) |
| Hydroxy CH. CHNOCl | −4.9982 | 1.593 (0.538) | (4.281, 15.971) |
| Anti-parasites | |||
| Aminoquinoline CHN | −5.5707 | (0.852) | (3.543, ) |
| Metrodinazole CHNO | −6.5734 | 2.034 (0.718) | (4.129, 14.665) |
| Amodiaquinine CHNOCl | −5.1099 | 1.259 (0.545) | (, 4.842) |
| Molecule/Weight | E (eV/atom) Molecule | E (eV/atom) Complex | Ads. Ene. (eV) Molecule |
|---|---|---|---|
| Industry | |||
| Biotinidase CHO/88.062 | 4.568 (4.580) | 1.230 | |
| Thymol CHO/150.218 | 4.497 (4.545) | 6.193 | 1.215 |
| Carbachol CHO/150.218 | 4.498 (4.544) | 6.192 | 1.163 |
| Glucose CHO/ 180.160 | 4.263 (4.334) | 6.152 | 1.706 |
| Gallic acid CHO/184.147 | 4.970 (5.070) | 6.462 | |
| Anthraquinone CHO/208.212 | (5.483) | 6.480 | 1.427 |
| Resveratrol CHNO/228.240 | 4.980 (5.023) | 6.272 | 1.029 |
| Molecule | E (eV) Complex | Gap(H-L) (eV) Complex | Electric Dipole (Dbys) Molecule/Complex |
|---|---|---|---|
| Industry | |||
| Biotinidasa CHO | −6.289 | (0.526) | (, ) |
| Thymol CHO | −5.127 | 2.246 (0.532) | (1.280, 7.136) |
| Carbachol CHO | −5.733 | 2.424 (0.571) | (1.190, 8.897) |
| Glucose CHO | −5.763 | 2.022 (0.401) | (3.270, 1.011) |
| Gallic acid CHO | −5.347 | 2.140 (0.662) | (3.658, 9.506) |
| Anthraquinone CHO | −6.372 | 1.792 (0.829) | (0.000, 11.731) |
| Resveratrol CHNO0 | −5.264 | 1.390 (0.531) | (1.694, 2.966) |
| Molecule | Ti-X (Distance) (Å) | X-C (Distance) (Å) | Ave. Ads. Energy (eV) |
|---|---|---|---|
| Binding by X | Ave. First Bond | Ave. Second Bond | |
| Vitamin | |||
| X=O | 2.07 (1.98–2.13) | 1.31 (1.25–1.47) | 1.577 (1.242, 1.996) |
| Amino acid | |||
| X=O | 2.07 (2.02–2.17) | 1.29 (1.25–1.42) | 1.590 (1.150, 1.953) |
| X=N | 2.16 (2.14–2.18) | 1.36 (1.36–1.37) | 1.961 (1.768, 2.062) |
| Analgesic | |||
| X=O | 2.07 (1.97–2.16) | 1.32 (1.27–1.50) | 1.667 (1.400, 1.936) |
| Antis- | |||
| X=O | 2.09 (2.02–2.20) | 1.31 (1.27–1.43) | 1.343 (1.097, 1.544) |
| X=N | 2.20 (2.18–2.24) | 1.38 (1.36–1.40) | 1.799 (1.561, 1.897) |
| Industry | |||
| X=O | 2.13 (2.04–2.20) | 1.37 (1.24–1.49) | 1.368 (1.029, 1.804) |
| Molecule | Total Energy | LE | HBond |
|---|---|---|---|
| Co-crystal | −8.73 (−201.31) | −0.175 (−4.11) | −0.132 (−3.05) |
| Prop8 | −11.09 (−255.79) | −0.236 (−5.44) | −0.346 (−7.99) |
| Fav | −3.21 (−74.00) | −0.292 (−6.73) | −0.046 (−1.06) |
| Clq | −5.86 (−135.02) | −0.266 (−6.14) | −0.075 (−1.74) |
| Hclq | −6.12 (−141.05) | −0.266 (−6.13) | −0.101 (−2.33) |
| Fav-(TiO) | −2.69 (−62.10) | −0.038 (−0.87) | −0.268 (−6.19) |
| Clq-(TiO) | −3.78 (−87.11) | −0.046 (−1.06) | −0.291 (−6.72) |
| Hclq-(TiO) | −3.65 (−84.16) | −0.044 (−1.01) | −0.327 (−7.55) |
| (TiO) | 0.30 (7.01) | 0.005 (0.12) | −0.409 (−9.43) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera-Granja, F.; Aguilera-del-Toro, R.H.; Díaz-Cervantes, E. Adsorption of Selected Molecules on (TiO2)20 Nano-Clusters: A Density-Functional-Theory Study. Nanomanufacturing 2022, 2, 124-145. https://doi.org/10.3390/nanomanufacturing2030010
Aguilera-Granja F, Aguilera-del-Toro RH, Díaz-Cervantes E. Adsorption of Selected Molecules on (TiO2)20 Nano-Clusters: A Density-Functional-Theory Study. Nanomanufacturing. 2022; 2(3):124-145. https://doi.org/10.3390/nanomanufacturing2030010
Chicago/Turabian StyleAguilera-Granja, Faustino, Rodrigo H. Aguilera-del-Toro, and Erik Díaz-Cervantes. 2022. "Adsorption of Selected Molecules on (TiO2)20 Nano-Clusters: A Density-Functional-Theory Study" Nanomanufacturing 2, no. 3: 124-145. https://doi.org/10.3390/nanomanufacturing2030010