
Effect of Vanadium Catalysts on Hydrogen Evolution from NaBH4


Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Equipment
2.3. Synthesis and Doping
3. Results
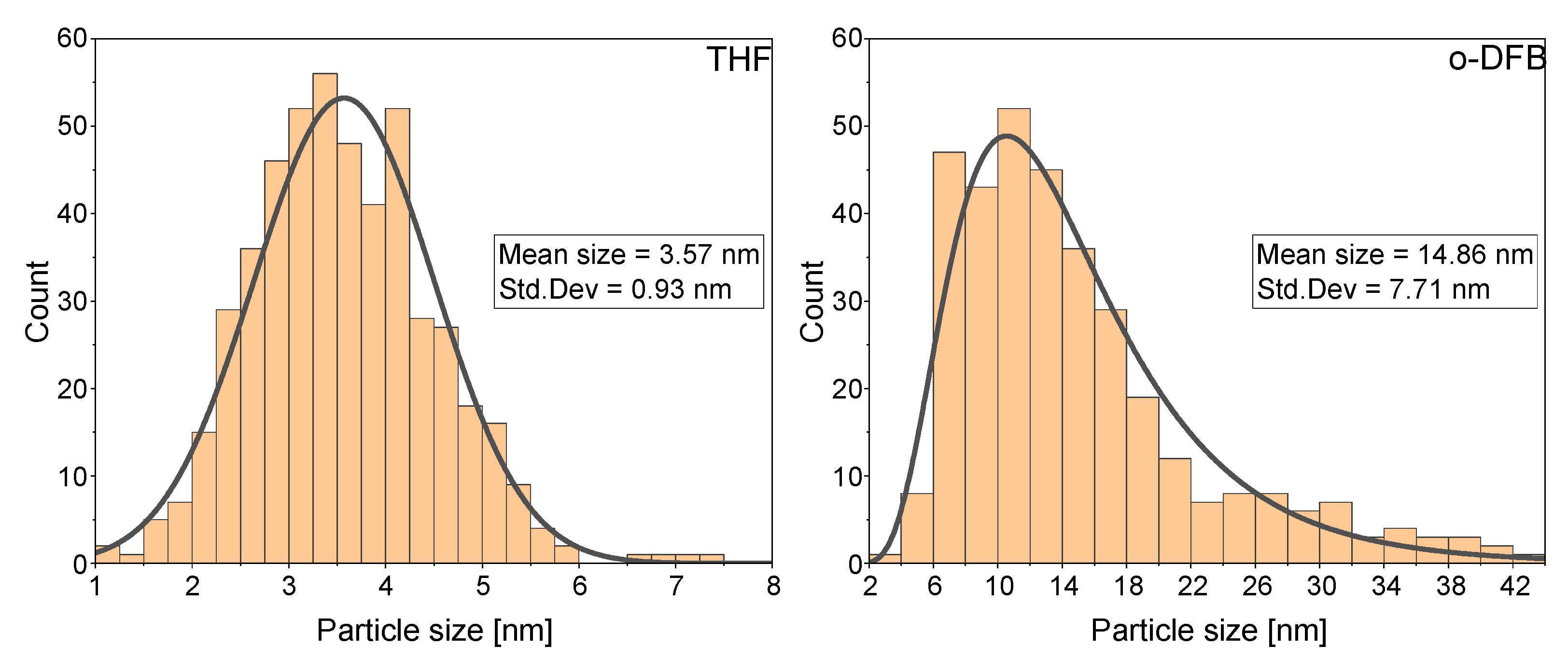
3.1. Synthesis of VNPs
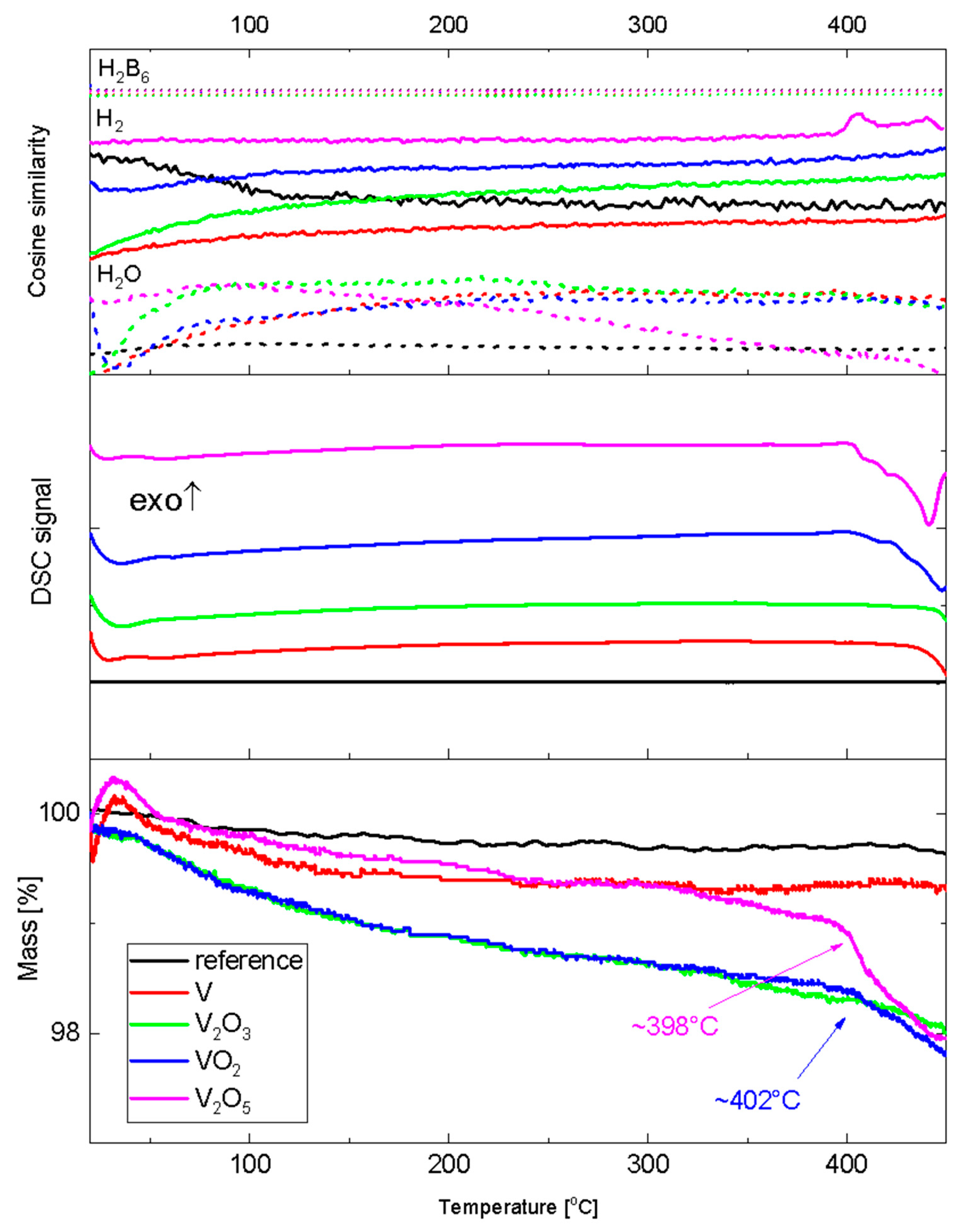
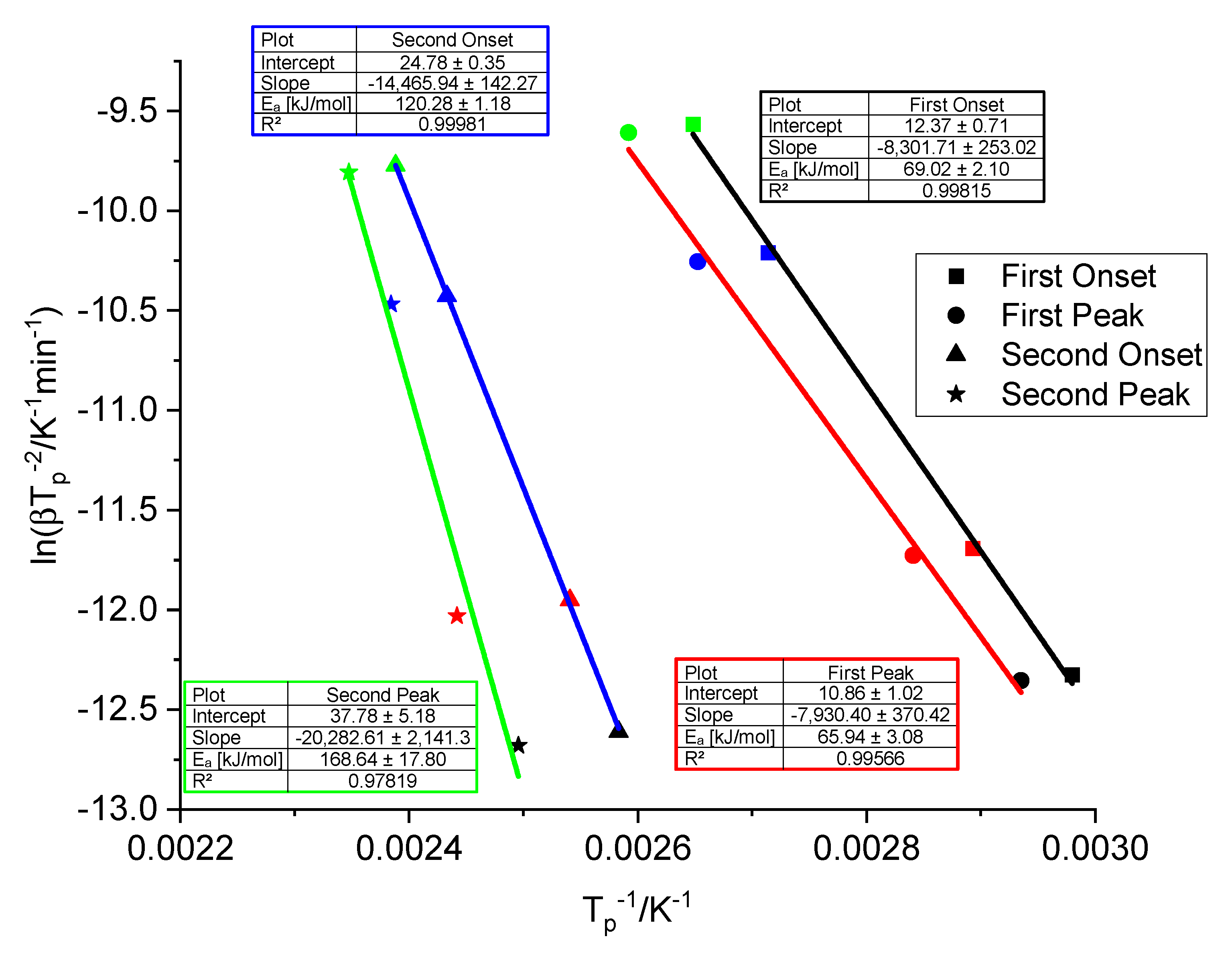
3.2. Thermogravimetric and Spectroscopic Analysis of Doped Samples
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grochala, W.; Edwards, P.P. Thermal decomposition of the non-interstitial hydrides for the storage and production of hydrogen. Chem. Rev. 2004, 104, 1283–1316. [Google Scholar] [CrossRef]
- Züttel, A. Hydrogen storage methods. Sci. Nat. 2004, 91, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Bannenberg, L.J.; Heere, M.; Benzidi, H.; Montero, J.; Dematteis, E.M.; Suwarno, S.; Jaroń, T.; Winny, M.; Orłowski, P.A.; Wegner, W.; et al. Metal (boro-) hydrides for high energy density storage and relevant emerging technologies. Int. J. Hydrogen Energy 2020, 45, 33687–33730. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. In Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group; World Scientific: Singapore, 2011; Volume 414, pp. 265–270. [Google Scholar]
- Graetz, J. New approaches to hydrogen storage. Chem. Soc. Rev. 2009, 38, 73–82. [Google Scholar] [CrossRef]
- Li, H.-W.; Zhu, M.; Buckley, C.; Jensen, T.R. Functional materials based on metal hydrides. Inorganics 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Mohring, R.M.; Eason, I.A.; Fennimore, K.A. Performance Bench Testing of Auotive-Scale Hydrogen on Demand™ Hydrogen Generation Technology; SAE Technical Paper: Warrendale, PA, USA, 2002. [Google Scholar]
- Huang, Y.; Cheng, Y.; Zhang, J. A Review of High Density Solid Hydrogen Storage Materials by Pyrolysis for Promising Mobile Applications. Ind. Eng. Chem. Res. 2021, 60, 2737–2771. [Google Scholar] [CrossRef]
- Martelli, P.; Caputo, R.; Remhof, A.; Mauron, P.; Borgschulte, A.; Zuttel, A. Stability and decomposition of NaBH4. J. Phys. Chem. C 2010, 114, 7173–7177. [Google Scholar] [CrossRef]
- Puszkiel, J.; Garroni, S.; Milanese, C.; Gennari, F.; Klassen, T.; Dornheim, M.; Pistidda, C. Tetrahydroborates: Development and potential as hydrogen storage medium. Inorganics 2017, 5, 74. [Google Scholar] [CrossRef] [Green Version]
- Milanese, C.; Jensen, T.R.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Yang, H.; Lombardo, L.; Zuettel, A.; Filinchuk, Y.; Ngene, P.; et al. Complex hydrides for energy storage. Int. J. Hydrogen Energy 2019, 44, 7860–7874. [Google Scholar] [CrossRef] [Green Version]
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Llamas, J.I.; Kalantzopoulos, G.N.; Nordholm, K.; Hauback, B.C. Destabilization of NaBH4 by transition metal fluorides. Molecules 2020, 25, 780. [Google Scholar] [CrossRef] [Green Version]
- Humphries, T.D.; Kalantzopoulos, G.N.; Llamas-Jansa, I.; Olsen, J.E.; Hauback, B.C. Reversible hydrogenation studies of NaBH4 milled with Ni-containing additives. J. Phys. Chem. C 2013, 117, 6060–6065. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Guo, Z.; Nevirkovets, I.P.; Liu, H.K.; Dou, S.X. Hydrogen de-/absorption improvement of NaBH4 catalyzed by titanium-based additives. J. Phys. Chem. C 2012, 116, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.F.; Yu, X.B.; Guo, Z.P.; Poh, C.K.; Liu, H.K.; Wu, Z.; Ni, J. Improvement of the LiAlH4–NaBH4 system for reversible hydrogen storage. J. Phys. Chem. C 2009, 113, 10813–10818. [Google Scholar] [CrossRef]
- Lai, Q.; Milanese, C.; Aguey-Zinsou, K.F. Stabilization of nanosized borohydrides for hydrogen storage: Suppressing the melting with TiCl3 doping. ACS Appl. Energy Mater. 2018, 1, 421–430. [Google Scholar] [CrossRef]
- Chong, L.; Zou, J.; Zeng, X.; Ding, W. Reversible hydrogen sorption in NaBH4 at lower temperatures. J. Mater. Chem. A 2013, 1, 13510–13523. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Wang, H.; Zhu, M. Hydrogen release from sodium borohydrides at low temperature by the addition of zinc fluoride. Int. J. Hydrogen Energy 2011, 36, 8203–8208. [Google Scholar] [CrossRef]
- Zou, J.; Li, L.; Zeng, X.; Ding, W. Reversible hydrogen storage in a 3NaBH4/YF3 composite. Int. J. Hydrogen Energy 2012, 37, 17118–17125. [Google Scholar] [CrossRef]
- Kumar, S.; Jaina, J.; Miyaoka, H.; Ichikawa, T.; Kojima, Y. Study on the thermal decomposition of NaBH4 catalyzed by ZrCl4. Int. J. Hydrogen. Energy 2017, 42, 22432–22437. [Google Scholar] [CrossRef]
- Huang, T.; Zou, J.; Zeng, X.; Wang, J.; Liu, H.; Ding, W. Reversible hydrogen sorption behaviors of the 3NaBH4-(x)YF3-(1-x)GdF3 system: The effect of double rare earth metal cations. Int. J. Hydrogen Energy 2019, 44, 4868–4877. [Google Scholar] [CrossRef]
- Chong, L.; Zou, J.; Zeng, X.; Ding, W. Effects of La fluoride and La hydride on the reversible hydrogen sorption behaviors of NaBH4: A comparative study. J. Mater. Chem. A 2014, 2, 8557–8570. [Google Scholar] [CrossRef]
- Chong, L.; Zou, J.; Zeng, X.; Ding, W. Mechanisms of reversible hydrogen storage in NaBH4 through NdF3 addition. J. Mater. Chem. A 2013, 1, 3983–3991. [Google Scholar] [CrossRef]
- Chong, L.; Zou, J.; Zeng, X.; Ding, W. Study on reversible hydrogen sorption behaviors of a 3NaBH4/HoF3 composite. Int. J. Hydrogen Energy 2014, 39, 14275–14281. [Google Scholar] [CrossRef]
- Afonso, G.; Bonakdarpour, A.; Wilkinson, D.P. Hydrogen storage properties of the destabilized 4NaBH4/5Mg2NiH4 composite system. J. Phys. Chem. C 2013, 117, 21105–21111. [Google Scholar] [CrossRef]
- Yahya, M.S.; Ali, N.A.; Sazelee, N.A.; Mustafa, N.S.; Yap, F.A.H.; Ismail, M. Intensive investigation on hydrogen storage properties and reaction mechanism of the NaBH4-Li3AlH6 destabilized system. Int. J. Hydrogen Energy 2019, 44, 21965–21978. [Google Scholar] [CrossRef]
- Nwakwuo, C.C.; Pistidda, C.; Dornheim, M.; Hutchison, J.L.; Sykes, J.M. Microstructural study of hydrogen desorption in 2NaBH4+MgH2 reactive hydride composite. Int. J. Hydrogen Energy 2012, 37, 2382–2387. [Google Scholar] [CrossRef]
- Garroni, S.; Pistidda, C.; Brunelli, M.; Vaughan, G.B.M.; Surinach, S.; Baro, M.D. Hydrogen desorption mechanism of 2NaBH4+MgH2 composite prepared by high-energy ball milling. Scr. Mater. 2009, 60, 1129–1132. [Google Scholar] [CrossRef]
- Mao, J.F.; Yu, X.B.; Guo, Z.P.; Liu, H.K.; Wu, Z.; Ni, J. Enhanced hydrogen storage performances of NaBH4–MgH2 system. J. Alloys Compd. 2009, 479, 619–623. [Google Scholar] [CrossRef]
- Mao, J.; Guo, Z.; Yu, X.; Liu, H. Improved hydrogen storage properties of NaBH4 destabilized by CaH2 and Ca(BH4)2. J. Phys. Chem. C 2011, 115, 9283–9290. [Google Scholar] [CrossRef]
- Somer, M.; Acar, S.; Koz, C.; Kokal, I.; Höhn, P.; Cardoso-Gil, R.; Aydemir, U.; Akselrud, L. Alpha-and beta-Na2[BH4][NH2]: Two modifications of a complex hydride in the system NaNH2–NaBH4; syntheses, crystal structures, thermal analyses, mass and vibrational spectra. J. Alloys Compd. 2010, 491, 98–105. [Google Scholar] [CrossRef]
- Pei, Z.; Wu, C.; Bai, Y.; Liu, X.; Wu, F. NaNH2–NaBH4 hydrogen storage composite materials synthesized via liquid phase ball-milling: Influence of Co–Ni–B catalyst on the dehydrogenation performances. Int. J. Hydrogen Energy 2017, 42, 14725–14733. [Google Scholar] [CrossRef]
- Mustafa, N.S.; Ismail, M. Significant effect of TiF3 on the performance of 2NaAlH4+ Ca(BH4)2 hydrogen storage properties. Int. J. Hydrogen Energy 2019, 44, 21979–21987. [Google Scholar] [CrossRef]
- Javadian, P.; Sheppard, D.A.; Buckley, C.E.; Jensen, T.R. Hydrogen storage properties of nanoconfined LiBH4–NaBH4. Int. J. Hydrogen Energy 2015, 40, 14916–14924. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Reed, D.; Paterakis, C.; Vasquez, L.C.; Baricco, M.; Book, D. Study of the decomposition of a 0.62LiBH4–0.38NaBH4 mixture. Int. J. Hydrogen Energy 2017, 42, 22480–22488. [Google Scholar] [CrossRef]
- Jensen, S.R.H.; Jepsen, L.H.; Skibsted, J.; Jensen, T.R. Phase Diagram for the NaBH4–KBH4 System and the Stability of a Na1–xKxBH4 Solid Solution. J. Phys. Chem. C 2015, 119, 27919–27929. [Google Scholar] [CrossRef]
- Ley, M.B.; Roedern, E.; Thygesen, P.M.M.; Jensen, T.R. Melting behavior and thermolysis of NaBH4−Mg(BH4)2 and NaBH4−Ca(BH4)2 composites. Energies 2015, 8, 2701–2713. [Google Scholar] [CrossRef] [Green Version]
- Dematteis, E.M.; Santoru, A.; Poletti, M.G.; Pistidda, C.; Klassen, T.; Dornheim, M.; Baricco, M. Phase stability and hydrogen desorption in a quinary equimolar mixture of light-metals borohydrides. Int. J. Hydrogen Energy 2018, 43, 16793–16803. [Google Scholar] [CrossRef]
- Fu, H.; Wu, Y.; Chen, J.; Wang, X.; Zheng, J.; Li, X. Promoted hydrogen release from alkali metal borohydrides in ionic liquids. Inorg. Chem. Front. 2016, 3, 1137–1145. [Google Scholar] [CrossRef]
- Xi, S.; Zhang, P.; Fu, Y.; Tome, K.C.; Zhang, T.; Zhou, S.; Guo, Y.; Yu, H. Hydrogen Release: Thermodynamic and kinetic studies of NaBH4 activated by different zeolite nanoparticles. Energy Fuels 2020, 34, 10218–10224. [Google Scholar] [CrossRef]
- Manoharan, K.; Palaniswamy, V.K.; Raman, K.; Sundaram, R. Investigation of solid state hydrogen storage performances of novel NaBH4/Ah-BN nanocomposite as hydrogen storage medium for fuel cell applications. J. Alloys Compd. 2021, 860, 158444. [Google Scholar] [CrossRef]
- Ampoumogli, A.; Steriotis, T.; Trikalitis, P.; Giasafaki, D.; Bardaji, E.G.; Fichtner, M.; Charalambopoulou, G. Nanostructured composites of mesoporous carbons and boranates as hydrogen storage materials. J. Alloys Compd. 2011, 509, S705–S708. [Google Scholar] [CrossRef]
- Ngene, P.; van den Berg, R.; Verkuijlen, M.H.W.; de Jong, K.P.; de Jongh, P.E. Reversibility of the hydrogen desorption from NaBH4 by confinement in nanoporous carbon. Energy Environ. Sci. 2011, 4, 4108–4115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xiao, X.; Fan, X.; Li, S.; Ge, H.; Wang, Q.; Chen, L. Fast hydrogen release under moderate conditions from NaBH4 destabilized by fluorographite. RSC Adv. 2014, 4, 2550–2556. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, J.; Hua, M.Y.; Zhou, D.W. Modifying effects and mechanisms of graphene on dehydrogenation properties of sodium borohydride. J. Mater. Sci. 2020, 55, 1959–1972. [Google Scholar] [CrossRef]
- Chong, L.; Zeng, X.; Ding, W.; Liu, D.; Zou, J. NaBH4 in “graphene wrapper:” Significantly enhanced hydrogen storage capacity and regenerability through nanoencapsulation. Adv. Mater. 2015, 27, 5070–5074. [Google Scholar] [CrossRef]
- Wang, T.; Aguey-Zinsou, K.F. “Surfactant-free” sodium borohydride nanoparticles with enhanced hydrogen desorption properties. ACS Appl. Energy Mater. 2020, 3, 9940–9949. [Google Scholar] [CrossRef]
- Wang, T.; Aguey-Zinsou, K.F. Controlling the growth of NaBH4 nanoparticles for hydrogen storage. Int. J. Hydrogen Energy 2020, 45, 2054–2067. [Google Scholar] [CrossRef]
- Christian, M.L.; Aguey-Zinsou, K.F. Core--shell strategy leading to high reversible hydrogen storage capacity for NaBH4. ACS Nano 2012, 6, 7739–7751. [Google Scholar] [CrossRef]
- Christian, M.; Aguey-Zinsou, K.F. Synthesis of core-shell NaBH4@M (M = Co, Cu, Fe, Ni, Sn) nanoparticles leading to various morphologies and hydrogen storage properties. Chem. Comm. 2013, 49, 6794–6796. [Google Scholar] [CrossRef]
- Li, Y.; Ding, X.; Zhang, Q. Self-printing on graphitic nanosheets with metal borohydride nanodots for hydrogen storage. Sci. Rep. 2016, 6, 31144. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lombardo, L.; Luo, W.; Kim, W.; Zuttel, A. Hydrogen storage properties of various carbon supported NaBH4 prepared via metathesis. Int. J. Hydrogen Energy 2018, 43, 7108–7116. [Google Scholar] [CrossRef]
- Maj, Ł.; Grochala, W. Theoretical design of catalysts for the heterolytic splitting of H2. Adv. Funct. Mater. 2006, 16, 2061–2076. [Google Scholar] [CrossRef]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B-N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.J.; Wiswall, R.H. Higher hydrides of vanadium and niobium. Inorg. Chem. 1970, 9, 1678–1682. [Google Scholar] [CrossRef]
- Koteras, K. Synteza i właściwości fizykochemiczne nowych, wysoce reaktywnych soli wapnia, tytanu i wanadu z anionami słabo koordynującymi. Master’s Thesis, Warsaw University of Technology, Warsaw, Poland, 2018. [Google Scholar]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Natl. Bur. Stand. 1956, 57, 217–221. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Wang, T.; Aguey-Zinsou, K.-F. Direct Synthesis of NaBH4 Nanoparticles from NaOCH3 for Hydrogen Storage. Energies 2019, 12, 4428. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sc [13] | Ti | V | Cr [13] | Mn [13] |
|---|---|---|---|---|
| ScF3—501 °C | Nano—493 °C [14] TiF3—300 °C [15] 5%mol TiF3—~300 °C [16] TiF4—522 °C [13] TiCl3 1:6—100 °C * [17] TiB2—503 °C [14] TiSiO4—489 °C [14] | VF4—499 °C [13] NaBH4 + PrF3 + VF3 3:1:0.2—417 °C [18] V3B—478 °C [14] | CrF3—500 °C | MnF3—483 °C |
| Fe [13] | Co | Ni | Cu [13] | Zn |
| FeF3—498 °C | CoF3—501 °C [13] Co3B—480 °C [14] | Nano—483 °C [14] NiF2—452 °C [13] NiF2—453 °C [14] wt.10% Ni on Si/Al2O3—449 °C [14] NiCl2—460 °C [14] Ni3B—462 °C [14] | CuF2—476 °C | ZnF2—504 °C [13] ZnF2 1:2—95 °C ** [19] |
| Y | Zr | Nb [13] | Rh [14] | Pd [14] | Ag [13] | Cd [13] |
|---|---|---|---|---|---|---|
| YF3—513 °C [13] YF3 3:1—423 °C [20] | ZrF4—503°C [13] ZrCl4—503 °C [21] | NbF5: 2%mol—442 °C 10%mol—379 °C 15%mol—379 °C | Rh (5 wt.%) on Al2O3—476 °C | Nano—420 °C | AgF—498 °C | CdF2—512 °C |
| (x)YF3 + (1 − x)GdF3 3:1 [22]: x = 2/3—449 °C x = 1/2—446 °C x = 1/3—440 °C |
| La [23] | Ce [13] | Pr [18] | Nd [24] | Ho [25] |
|---|---|---|---|---|
| LaF3 3:1—396 °C La 3:1—442 °C | CeF3—506 °C CeF4—502 °C | PrF3 3:1—439 °C | NdF3 3:1—413 °C | HoF3 3:1—443 °C |
| MH2 | Mg2NiH4 [26] | NaNH2 | M(AlH4)x | Li3AlH6 [27] | MBH4 |
|---|---|---|---|---|---|
| M = Mg: 1:2—420 °C [28] 1:2—490 °C [29] 1:2 with 5%mol of [30]: TiF3—~470 °C TiO2—~470 °C Zr—~470 °C Si—~470 °C BCC—~470 °C M = Ca: 1:6—390 °C [31] | 5:4—~350 °C | 1:1—330 °C * [32] 2:1 with Co–Ni–B catalyst [33]: 1 wt.%—~300 °C 3 wt.%—~285 °C 5 wt.%—285 °C 7 wt.%—~285 °C | M = Li: 1:1 [16]: No catalyst—446 °C 5% TiF3—~300 °C M = Ca: 2NaAlH4 + Ca(BH4)2 with 5 wt.% TiF3 —400 °C [34] | 1:1 (1 h)—400 °C 1:1 (24 h)—392 °C 1:2—430 °C 1:3—430 °C | M = Li 0.38:0.62: pure—350 °C [35] pure—~420 °C [36] 60%vol in carbon areogel—~333 °C [35] M = K—~472 °C [37] M = Mg—~205 °C [38] M = Li, K, Mg, and Ca (equimolar)—~330 °C [39] |
| Ionic Liquids [40] | Carbon | Carbon Based | Zeolites [41] | Ah-BN [42] | NaBF4 ** [34] |
|---|---|---|---|---|---|
| 1%mol solution in: bmimBr—~165 °C bmimNTf2—104 °C | 20% of scaffolds CMK-3 —235 °C and 380 °C * [43] ~25% of HSAG-500 [44]: infiltrated—~270 °C melted—~300 °C mixed—~320 °C | 45% fluorographite—141 °C [45] graphene: 10%—370 °C [46] ?—426 °C [47] | MCM-22: 1:1—490 °C 1:2—503 °C 1:3—507 °C 1:4—508 °C SAPO-34: 1:1—483 °C 1:2—491 °C 1:3—493 °C 1:4—495 °C | 1:1—399 °C | 1:10 mol—468 °C 1:2 mol—305 °C |
| Solvented Ion Stabilization * [48] | Evaporation with 10%mol of Ligands or Solvents [49] | Anti-Solvent Precipitation | Milled LiBH4 + NaCl |
|---|---|---|---|
| LiCl—495 °C MgCl2—445 °C NaI—485 °C | Hexyloamine—489 °C dodecyloamine—476 °C octodecyloamine—473 °C tertabuthylamonium bromide—464 and 482 °C tetraoctylamonium bromide—484 °C tetradecyloamonium bromide—~500 °C dodecane—478 °C dodecanothiol—464 °C tri-decylic acid—~400 and 474 °C tetrabutylphosphonium bromide—471 °C | Bare—460 and 535 °C [50] Coated with: Ni—418 °C [50] Co—350 °C [51] Cu—400 °C [51] Fe—380 °C [51] Sn—450 °C [51] | Graphene coated (ultrasonicated and dried)—400 °C [47] Milled with graphene nanoflakes—~300 °C [52] Milled 1:10 mass with [53]: graphite—109 °C graphene oxide—114 °C carbon nanotubes—116 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orłowski, P.A.; Grochala, W. Effect of Vanadium Catalysts on Hydrogen Evolution from NaBH4. Solids 2022, 3, 295-310. https://doi.org/10.3390/solids3020021
Orłowski PA, Grochala W. Effect of Vanadium Catalysts on Hydrogen Evolution from NaBH4. Solids. 2022; 3(2):295-310. https://doi.org/10.3390/solids3020021
Chicago/Turabian StyleOrłowski, Piotr Antoni, and Wojciech Grochala. 2022. "Effect of Vanadium Catalysts on Hydrogen Evolution from NaBH4" Solids 3, no. 2: 295-310. https://doi.org/10.3390/solids3020021