

The Cross-Talk between Microbiome and Metabolome in Rheumatoid Arthritis
by
 and
and
Lidia La Barbera
1,†,
Chiara Rizzo
1,†,
Giulia Grasso
1,
Federica Macaluso
2,
Federica Camarda
1,
Francesco Ciccia
3 and
Giuliana Guggino
1,* 1
Rheumatology Section, Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties, University of Palermo, Piazza delle Cliniche 2, 90110 Palermo, Italy
2
Rheumatology Unit, Department of Internal Medicine, University of Modena and Reggio Emilia, AUSL-IRCCS, Via Giovanni Amendola, 2, 42122 Reggio Emilia, Italy
3
Division of Rheumatology, Department of Precision Medicine, University of Campania Luigi Vanvitelli, S. Andrea delle Dame—Via L. De Crecchio 7, 0138 Naples, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
BioChem 2023, 3(1), 47-60; https://doi.org/10.3390/biochem3010004
Submission received: 31 January 2023
/
Revised: 26 February 2023
/
Accepted: 6 March 2023
/
Published: 13 March 2023
Abstract
:Modern “omics” sciences, including metabolomics and microbiomics, are currently being applied to inflammatory autoimmune diseases, such as rheumatoid arthritis (RA), to investigate the interplay between microbiota, metabolic function, and the immune system. In recent decades, robust evidence has suggested that disruption of the normal composition of the microbiome, known as dysbiosis, in the gut and mouth of RA patients contributes to immune dysregulation and alterations in the metabolic pathways, shaping the pathogenesis of the disease and playing a central role in the risk and progression of RA. Metabolic pathways can be influenced by various agents such as the surrounding environment, lifestyle, and exposure to microbiota imbalance. In turn, the body’s metabolic homeostasis influences the immune response, making metabolomics helpful not only to understand pathogenesis pathways, but also to improve early disease detection and therapeutic chances. Combined gut microbiome and metabolome studies set out to unravel the interactions between these two entities, providing insights to discover new treatment targets and potential biomarkers to prevent joint damage. The purpose of this review is to summarize the main recent findings that suggest promising new research directions for the pathogenesis of RA.
1. Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease, clinically characterized by poly-articular involvement with chronic synovial inflammation culminating in bone erosions and disability [1].
RA is considered a multistep disease [2]. In the preclinical phase of the disease, genetic predisposition and environmental triggers increase susceptibility to the onset of RA, resulting in the activation of the immune system outside the joint compartment, mainly in the mucosal surfaces. Despite the typical joint involvement in established RA, the pathogenesis of the disease likely begins far from joint structures, in the lungs, periodontium, or intestine [3]. In a second phase, the immune response is amplified systemically, with an expansion of the autoantibody repertoire. Finally, the synovial compartment becomes the target of a local inflammatory response that culminates in clinically evident synovitis [4].
Growing evidence has proposed gut microbiome dysbiosis as a trigger environmental factor for the dysregulation of innate and adaptive immune responses and the onset of RA.
The suggested mechanism for the gut–joint inflammatory axis is related to intestinal leakage, which can expose the immune system to microorganisms and their products, triggering a systemic immune response resulting in a local inflammatory process within the joints.
In the RA scenario, the loss of immunological tolerance and disease development are related events. The immunological mechanisms that lead to cell differentiation and expansion both depend on antigen recognition and activation of metabolic pathways, which provide cellular energy supply [5].
Early stages of the disease involve innate immune cells, such as monocytes and dendritic cells (DCs), that orchestrate the inflammatory cascade, which subsequently leads to the activation of lymphocytes and autoantibodies’ production [6]. Their activation is stimulated by Toll-like receptors (TLR) that bind antigens, driving and supporting pathogenetic mechanisms through their role as antigen-presenting cells, phagocytes, and chemokine producers [7].
In particular, macrophages are abundant in RA synovium and represent the major source of lytic enzymes, chemokines, and cytokines, leading to joint damage and neoangiogenesis. In addition, they exhibit a long-lasting immune memory capable of remodulating when stimulated [8]. Naive T lymphocytes in the RA inflammatory milieu age quickly, and drive cellular metabolic reprogramming [5] and macrophage activation.
Similarly, chronic inflammation in RA appears to be related to metabolic alterations, as confirmed by the finding of some metabolites linked to rapid disease progression, although they may precede the onset of symptoms by many years.
Combined studies on immune cells, gut microbiome, and metabolome aim to investigate the interactions between these entities, providing new insights for the improvement of therapeutic strategies and for the identification of potential biomarkers in order to provide new tools to interfere with the disruption of the immunological homeostasis that drives RA, in order to prevent disability and joint damage.
Nowadays, the identification of a specific biomarker or a panel of molecules to profile RA patients, especially in the initial stage of disease or in the case of seronegative disease, is a need that has still not been met in rheumatology. The lag time between symptom onset and diagnosis is up to 9–17 months [9], meaning that patients may experience the risk of losing the so-called ‘window of opportunity’ for treatment initiation that lasts about 3 months after first symptoms and has been identified as the optimum time to achieve remission [10].
In this regard, the development of sequencing technologies and multi “omics” methodologies may allow a better characterization of RA to help risk prediction, early diagnosis, prognosis, and proper monitoring.
Over the last decade, thanks to metabolomics, a deeper description of the metabolic profile, at both serum and tissue level, of RA has emerged. The metabolic fingerprinting in such a disease is a valuable tool to address specific questions regarding the link between dysbiosis, bacterial metabolites, and disease development in order to acquire more knowledge on RA pathogenesis and management [11].
Therefore, the purpose of the present review is to dissect the current literature regarding the interaction between the microbiome, and specifically gut dysbiosis, and RA through the information derived from metabolomic studies, which stand out as one of the most promising high-precision techniques in rheumatology.
Research Methodology
A literature search was performed with the aim of achieving a comprehensive and structured analysis of studies. We searched the major databases (MEDLINE, EMBASE, ScienceDirect, PubMed, Scopus) to identify relevant articles for inclusion. Two reviewers independently screened the literature search results and abstracted data from included studies. A systematic search was performed using keywords related to the diseases of interest (RA) and the main topics (microbiome, dysbiosis, and metabolome). The reference lists of all included articles and recent reviews were evaluated for their relevance to disease pathogenesis and their impact on outcome prediction and disease management.
2. Metabolome in RA
Metabolomics is an emerging science that is part of the omics group (e.g., proteomic, transcriptomics, etc.). It allows the identification of small molecules, known as metabolites, in a biologic system, catching the alteration of the metabolic status of different tissues and fluids that mirror the cellular perturbation occurring during disease.
Metabolic pathways can be influenced by various agents such as the surrounding environment, lifestyle, and exposure to microbiota imbalance. In this regard, links between diet-related metabolites and changes in the microbiome have been extensively investigated [12]. In turn, the body’s metabolic homeostasis influences the immune response [13], making metabolomics helpful not only to understand pathogenesis pathways, but also to improve early disease detection and therapeutic chances [13].
Nowadays, in the “omics era”, numerous metabolomics assays of peripheral blood, tissues, and urine in patients with RA have been conducted, and many techniques have been employed to characterize metabolomics. Metabolomics is based on two main high-throughput technologies: nuclear magnetic resonance (NMR) and mass spectrometry (MS). After the acquisition of experimental data, multivariate statistical analysis is performed in order to identify the patient’s metabolic profile. MS is more sensitive, but requires sample pre-treatment through separation of individual constituents using liquid chromatography or gas chromatography; this results in greater variability and lower reproducibility. In contrast, NMR does not require sample pre-treatment, yielding more reproducible NMR despite lower sensitivity. In both cases, a plot is obtained that identifies the different metabolites representing the metabolome of a specific sample [14].
Metabolomics has been applied in various fields of medicine, as it has enabled the identification of new possible biomarkers through new methods. This justifies the growing interest in its application in the study of rheumatic diseases.
A recent study demonstrated a correlation between inflammation in the early stages of immune diseases, as detected using C reactive protein (CRP) levels, and the serum/urinary metabolome. The most abundant metabolites present in the samples included glucose, amino acids, lactate, and citrate [15]. In 2013, Young et al. showed a relationship between the early inflammatory stages of RA and the levels of specific metabolites such as low-density lipoproteins, lipids, lactate, glucose, methylguanidine, amino acids, and their derivatives [16]. Nevertheless, inflammation has been related to metabolites derived from oxidative stress processes, the urea cycle, and catabolism protein [17], raising the profile of the urinary metabolome, which, so far, has been studied mainly to evaluate and predict pharmacological response [18] and to accelerate the diagnostic process [19].
However, no differences in metabolites were found between RA patients in the early stage and advanced stage, or between the seropositive and seronegative forms.
Interestingly, a negative correlation was identified between the levels of CRP and citrate, which can be explained by the metabolic reprogramming process resulting from the activation of the immune response [20]. Once innate immune cells such as macrophages and DCs are activated, the glycolysis and pentose phosphate pathways are upregulated, while the citrate pathway, oxidative phosphorylation, and fat acid oxidation are reduced. However, the increase in glycolytic flux can lead to an accumulation of citrate and succinate in the mitochondria, which, once transported to the cytosol, is broken down to form substances necessary for the activation of macrophages and DCs [21].
Conversely, a positive correlation between CRP levels and succinate was highlighted. This finding is consistent with the ability of succinate to stimulate IL-1ß production during the inflammatory process and, furthermore, post-translational processes such as succinylation are able to sustain inflammation [22].
Despite the positive association with glucose and lactate, serum CRP levels correlated negatively with pyruvate levels. Several mechanisms could justify elevated glucose levels during an inflammatory response in order to satisfy the increased cellular demand [23]. Mainly, the activation of glycolysis and the reduction of pyruvate to lactate are both pathways required for the perpetuation of the immune response [24].
Since the establishment of the inflammatory response requires adequate bioavailability of metabolites to meet the cellular demand necessary for clonal expansion and cytokine production, some metabolites are, therefore, considered potential biomarkers that can predict the onset of the disease and, simultaneously, potential therapeutic targets [5].
However, it should be considered that the major cell lines adapt to different metabolic conditions: T cells reduce glycolysis and prefer anabolic reactions, while macrophages, on the other hand, store glucose in favor of cytokine production. The reason why major cell lines rely on different pathways could be related to the reprogramming of immune cells, influenced by the inflammatory environment.
The different metabolic adaptations mirror the main roles of the involved cell lines. T cells mainly perform a regulatory and memory function. Macrophages predominantly act as effector cells [25]. Anabolic reactions seem to be the exclusive prerogative of T lymphocytes, in particular of naive T lymphocytes and memory cells, while macrophages have developed an adaptation in a hypermetabolic sense and this imprinting is already present in undifferentiated monocytes [5].
Metabolic signature can predict arthritis onset in its early stages and can influence autoantibodies’ production, also according to genetic susceptibility.
Studies of genome-wide association (GWAS) highlighted the presence of many loci associated with metabolite levels, which are involved in various metabolic pathways, indicating the genetic influence on the metabolome. These pathways include amino acids, lipid metabolism, carnitines, fatty acid metabolism, intermediates of purine and pyrimidine metabolism, glucose homeostasis, vitamin, and cofactor levels; the genetic polymorphisms of genes linked to these metabolic pathways in RA have been described [26,27].
Specifically, GWAS studies have identified approximately 100 genetic loci implicated in the etiopathogenesis of RA [28].
3. Microbiome in RA
Beyond the genetic susceptibility, which is also strongly related to the presence of human leukocyte antigen (HLA)-DRB1 shared epitope (HLA-SE) alleles, great interest has developed around other environmental factors that may trigger the autoimmune process in RA (Figure 1) [29]. The underlying inflammatory and autoimmune mechanisms are triggered approximately one decade before RA clinical onset, resulting in the detection of anti-citrullinated protein antibodies (ACPA) at different sites during the pre-clinical phase of the disease [30].
Numerous studies on microbiota composition and on the different expression of its metabolites have been conducted, highlighting a correlation between RA etiopathogenesis and microbiome imbalance at multiple levels [31,32,33]. Disease development could be prompted by a regulatory mechanism induced by the microbiota and its metabolites, which are able to drive the immune response and the inflammatory process [34]. Specifically, the process of protein citrullination mediated by Porphiromonas gingivalis (P. gingivalis) and the activation of T helper (Th) 17 pathways by Prevotella copri (P. copri) are recognized among the mechanisms underlying the aberrant immune responses occurring during disease [35,36].
Several studies have reported the increased risk of developing RA in patients affected by periodontitis. The involvement of the bacteria coming from the oral cavity has an important role and influence on the immunity response in RA. The well-established link between periodontitis and RA is defined by the colonization of the oral cavity by the Gram-negative bacterium P. gingivalis [37]. P. gingivalis is involved in the production of citrullinated proteins through its enzyme P. gingivalis peptidyl-arginine deiminase (PPAD), which is able to convert arginine residues to citrulline [38]. Mechanistic studies in animal models have revealed that this periodontal bacterium significantly worsens the severity of collagen-induced arthritis (CIA) by the activation of the Th17-related pathway [39].
Recently, an alternative periodontal pathogen, Aggregatibacter actinomycetemcomitans, has been identified as trigger of autoimmunity in RA [40]. Its virulence factor, leukotoxin A (LtxA), may cause hypercitrullination of neutrophils in RA and, thus, allow the bacteria to evade host immune defenses [41]. Looh et al. [42] hypothesized that LtxA-mediated hypercitrullination may involve the abnormal activation of PAD enzymes, which leads to the citrullination of proteins and autoantigens recognized by ACPA. These mechanisms can further accelerate the development of RA in susceptible individuals.
RA susceptibility is not only influenced by oral microbiota, but also by gut dysbiosis, as demonstrated in multiple animal and human studies (Table 1). Xiaofei Liu et al., through 16S rRNA sequencing, characterized the intestinal microbiome in DBA1 mice that did or did not develop CIA [43]. In CIA-susceptible mice, they observed an enrichment of operational taxonomic units (OTUs) affiliated with the genus Lactobacillus before the arthritis onset and an increase in OTUs affiliated with the families Bacteroidaceae, Lachnospiraceae, and S24-7 after the disease development.
Strikingly, the authors described a higher frequency of arthritis induction in germ-free (GF) mice conventionalized with the microbiota from CIA-susceptible mice compared to those conventionalized with the microbiota from CIA-resistant mice. Finally, they also reported higher serum concentration of interleukin (IL)-17 and increased proportions of CD8+ T cells and Th17 lymphocytes in the spleen of CIA-susceptible mice [43].
In an elegant study, Chen Y. et al. analyzed the composition of the gut microbiome in RA patients using 16S rRNA gene sequencing, and identified different fecal metabolites, suggesting that alterations in fecal microbiota may result in alterations in fecal metabolites. The results of this analysis showed that the RA microbiome, as well as metabolites, differed substantially from those of healthy controls (HCs). Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria were the principal components in the gut microbiota in both groups, but they differed mainly in abundance and not in composition [44].
The causes of altered gut flora include the reduction in probiotics, such as Bifidobacterium, and the increase in dangerous species. Such disruption would affect different metabolic pathways, leading to the onset of disease. However, some controversial results have been described.
For instance, although the presence of Bifidobacterium was constantly reduced, Lactobacillus was more abundant in HCs in one study, while it was overexpressed in the RA group in another report [37]. This suggests that the probiotic imbalance could follow the different stages of RA [45].
In addition, some peptides (Val Thr Ile, Ile Gly Gly Ile, Val Asn Ile, etc.), amino acids (lysine, methionine), and nucleotides (thymidine, deoxyuridine, deoxyinosine, deoxyguanosine, etc.) were overexpressed in the RA group, whereas some lipids were decreased. Two fatty acids metabolites were identified as potential biomarkers; specifically, long-chain fatty acids (9,12-octadecadiynoic acid and 10Z-nonadecenoic acid) were increased in the RA group and were related to the presence of Verrucomicrobia and Akkermansia, suggesting that these two metabolites can influence the disease development [33].
These results confirm what has already been demonstrated in other studies in which fatty acids were increased in RA patients and their presence was related to the inflammatory process [46]. Consistently, some metabolic pathways, including the biosynthesis process of fatty acids and the degradation of glycosaminoglycans, were increased in subjects with RA compared to HCs [47]. Some long-chain fatty acids have shown a protective role, improving symptoms of RA, while short-chain fatty acids (SCFAs), the products of microbial metabolism, have a pivotal role in the gut–joint axis in animal models [48,49,50]. Several factors contribute to the maintenance of intestinal homeostasis, mainly SCFAs, followed by IgA secreted by plasma cells in the lamina propria, α-defensins derived from Paneth cells, polysaccharide A from the commensal Bacteroides fragilis (B. fragilis), and RegIIIγ, an antibacterial lectin expressed by epithelial cells [50]. Disruption of this delicate balance can result in the development of chronic inflammatory diseases, including RA. SCFAs, mainly butyrate, propionate, and acetate, are derived from the bacterial fermentation of non-digestible carbohydrates such as fiber; they act as histone deacetylase (HDAC) inhibitors and G- protein-coupled receptor (GPCR) ligands modulating the function of immune cells in the gut and other tissues [51]. Furthermore, SCFAs, as active microbial metabolites, regulate intestinal homeostasis by facilitating the assembly of tight junctions and the production of antimicrobial peptides. SCFAs can also affect drug response, as demonstrated by the role of butyrate in the beneficial effect of B. fragilis on the efficacy of therapy with methotrexate (MTX). When added to drinking water during MTX treatment, butyrate restored the therapeutic effect of MTX in gut microbiota-deficient CIA mice to a similar level as that achieved with B. fragilis gavage [52]. These findings further support a potential microbial intervention strategy to improve the management of RA.
Another study showed the development of adjuvant-induced arthritis (AIA) only in GF rats, whereas the animals colonized with Gram-negative bacteria, such as Escherichia coli or Bacteroides, showed only a mild phenotype of arthritis [53]. The same group has also described a protective role of some Gram-negative Enterobacteria (Escherichia coli and Bacteroides species) when introduced into otherwise GF arthritis-susceptible rats [54], supporting the theory of bacterial modulation in disease onset and development [55].
In humans, although it has been observed that a lower risk of developing RA is associated with recent gastrointestinal and/or urogenital infections [56], there is a substantial body of evidence supporting the association between RA and intestinal dysbiosis.
Several studies have reported alterations of the intestinal epithelial barrier and increased gut permeability in RA patients, especially linked with the intake of non-steroid anti-inflammatory drugs (NSAIDs) and the severity of disease activity [57,58,59,60,61].
Additionally, it has been proven that a reduction in gut microbial diversity occurs in RA patients compared to HCs, characterized by an abundance of some rare taxa, such as Actinobacteria. Moreover, an association between the increase in Collinsella, Eggerthella, and Faecalibacterium genera and the production of IL-17A has been described [32].
Zhang et al. investigated the oral and gut microbial community of RA patients and controls, detecting a dysbiosis at both sites in RA patients, partially attenuated after treatment with disease-modifying antirheumatic drugs (DMARDs) [62].
In particular, they described a rise in Gram-positive bacteria and a depletion of Gram-negative bacteria, such as Proteobacteria and Firmicutes, in the gut of RA patients. Moreover, at all sites, in individuals with RA, they observed a reduction in Haemophilus spp., negatively correlated with titers of the RA-specific autoantibodies, and an increase in Lactobacillus salivarius, associated with high disease activity.
In an interesting study, Scher et al. characterized and compared intestinal microbiota between new-onset DMARDs/steroids naive RA patients (NORA), chronic treated RA patients, psoriatic arthritis (PsA) patients, and HCs [63]. They performed sequencing of the 16S gene on stool samples, identifying higher levels of P. copri associated with decreased commensal beneficial Bacteriodes in NORA patients. Additionally, they highlighted the capacity of P. copri to regulate the gut microbiota and to induce colitis in an inflammatory bowel disease (IBD) mice model.
A recent study has also shown a marked sequence homology between two autoantigens, N-acetylglucosamine-6-sulfatase (GNS) and filamin A (FLNA), that are highly expressed in the synovial tissue of RA patients, and epitopes from Prevotella sp. This result further supports the molecular mimicry theory in RA and the association between mucosal and joint inflammation [64].
4. Metabolomics Applied to Treatment and Interference with Microbiome Pathological Dysfunction in RA
The metabolic profile of RA patients changes importantly during disease course, as reviewed above, but treatments could even contribute to altering metabolite levels, for example increasing anti-inflammatory molecules. In this regard, metabolomics could help in identifying new biomarkers to assess response to treatment with the final goal of profiling responders and non-responders, and of creating a more personalized approach to prevent treatment failure [65,66].
Currently, a large proportion of patients still shows a poor response to both conventional (c)DMARDs and biologic (b)DMARDs [67].
Regarding cDMARDs, the metabolome of RA patients revealed that the levels of tryptophan are decreased under treatment with MTX or leflunomide [68]. Tryptophan is involved in the metabolic pathway that drives TLR activation, danger molecule recognition, and IL-6 production, so its reduction could mirror the efficacy of MTX [69,70]. In an early RA cohort of 82 patients treated with MTX, a recent study demonstrated that the baseline concentrations of homocysteine, taurine, adenosine triphosphate, guanosine diphosphate, and uric acid were significantly lower in the plasma of non-responders versus responders, while glycolytic intermediates 1,3-/2,3-diphosphoglyceric acid, glycerol-3-phosphate, and phosphoenolpyruvate were significantly higher in non-responders [71]. In addition, in a mouse model of RA, low serum levels of N-methylisoleucine were revealed as a potential biomarker of response to MTX [72]. The effects of MTX even include the normalization of taurine, aspartate, alanine, lactic acid, adenosine, and guanine, whose levels are reestablished after treatment [73].
In relation to gut microbiome, Artacho et al. reported a possible association between gut bacteria and response to MTX in new-onset naive RA. In non-responders, gut bacteria showed an increased metabolism of MTX, possibly accounting for a decrease in drug availability for patients, leading to a worsened clinical response [74].
Among biologic agents, most evidence linking metabolomics to treatment focused on TNF-inhibitors (TNFi), as TNF is a strong proinflammatory cytokine renowned to influence glucose and lipid metabolism [75]. In particular, an increase in hippuric acid, citrate, and lactic acid was described after infliximab use, while an increase in choline, phenylacetic acid, urea, creatine, and methylamine was observed after treatment with etanercept [76].
A specific metabolite signature was even outlined between responders and non-responders to TNFi both at the blood and urine level. After TNFi therapy, two different metabolic profiles differentiated good responders from non-responders, and carbohydrate derivates, such as D-glucose, D-fructose, sucrose, and maltose, emerged as determinants of the therapeutic response [77]. Additionally, five metabolites (glycerol 3-phosphate, betonicine, N-acetylalanine, hexanoic acid, and taurine) were identified as potential predictors of good response to TNFi.
In urine samples, histamine, glutamine, phenylacetic acid, xanthine, xanthurenic acid, and creatinine were found to be upregulated in patients responding to TNF antagonist, while ethan-olamine, hydroxyphenylpyruvic acid, and phosphocreatine were downregulated.
Three metabolites, namely 3-aminobutyric acid, citric acid, and quinic acid, were shown to positively predict response to abatacept [78].
Changes in arachidonic acid metabolism with the consequent modulation of the inflammatory pathway related to IL-6 signaling were depicted in a study focused on tocilizumab efficacy [79]. Moreover, tocilizumab responders present higher levels of 3-hydroxybutyrate and phenylalanine, which can improve the ability to predict tocilizumab responders [80].
Rituximab administration determines a downregulation in phosphatidylethanolamines, phosphatidyserines, and phosphatidylglycerols levels, once again stressing the im-portance of amino acids and lipid perturbations in RA [81,82].
Coming to new treatments, specifically JAK-inhibitors, higher levels of omega-3 fatty acids and docosahexaenoic acid in treated patients were demonstrated and correlated with an important effect on pain reduction [83].
To improve the identification of possible responders to treatments, the combination of metabolites and clinical data is crucial [84], and new players, such as gut bacteria and alternative therapies targeting their functions, should carefully be taken into account.
On the basis of substantial evidence regarding the correlation between gut dysbiosis and RA, the effect of probiotics in RA patients has been recently investigated, with some encouraging results. Probiotics are live bacteria that, when administered to the host in appropriate doses, provide significant health benefits. Specifically, exogenous bacteria can improve gut microbiota homeostasis, modulate the immune response, and support gut barrier integrity, treating dysbiosis and related pathological conditions. Several studies have investigated the role of probiotic bacteria in systemic immune responses. In these studies, the supplementation with probiotics correlated with a decrease in disease activity score (DAS28), CRP, and systemic inflammatory cytokines, such as tumor necrosis factor-α (TNFα), IL-12, and IL-6, and at the same time, with an increase in IL-10 [45,85]. To identify potential probiotic gut microbes that can ameliorate the development of RA, microbiota profiling in RA patients was studied using 16S rDNA bacterial gene sequencing and shotgun metagenomics. Interestingly, the relative abundance of Parabacteroides distasonis (P. distasonis) in RA patients was decreased, and this decrease was negatively correlated with DAS28. These findings were explained by the effects of microbial metabolites derived from P. distasonis, lithocholic acid (LCA), deoxycholic acid (DCA), isolithocholic acid (isoLCA), and 3-oxolithocholic acid (3-oxoLCA), on CD4+ T-cell differentiation and macrophage polarization. In particular, 3-oxoLCA and isoLCA promoted the M2 polarization of macrophages and, simultaneously, inhibited Th17 cell differentiation. Consistently, oral treatment of arthritic mice with live P. distasonis significantly mitigated RA development, pointing to P. distasonis as a potential probiotic agent in the treatment of RA [86]. Another potential probiotic agent suggested for RA is Bifidobacterium pseudocatenulatum, which is also involved in bile acid (BA) metabolism. B. pseudocatenulatum prevented joint damage in a CIA mouse model by elevating the enzymatic activity of bile salt hydrolase and increasing the levels of unconjugated secondary BA to suppress aberrant Th 1/17-type immune responses [87]. Probiotics have been proposed as an adjuvant treatment in the therapeutic strategy of immune-mediated disorders. MTX, one of the most widely used DMARDs in inflammatory arthritis, alters the composition of the gut microbiota, helping to restore the altered microbial balance [62]. Further introduction of a probiotic has demonstrated a synergistic effect that can effectively control the inflammatory status. In this regard, the positive influence of the Lactobacillaceae family on the microbiota in RA has been optimized in patients taking DMARDs, as demonstrated by the ability of Lactobacillus casei (L. casei) to reduce RA symptoms and inhibit proinflammatory cytokines [88]. In their study, Alipour et al. showed that L. casei supplementations decreased serum CRP levels, and reduced tender and swollen joint counts; furthermore, serum levels of interleukins (IL-10, IL-12) and TNF-α were improved in the probiotic group [88]. The impact of lactobacilli on joint inflammation has also been explored in the CIA model. The addition of L. casei or L. acidophilus in a rat model of CIA proved to be as effective as standard treatment with indomethacin [89]; the anti-inflammatory properties of L. casei in the CIA rat model were confirmed by its ability to inhibit the COX-2 enzyme and downregulate proinflammatory cytokines [90]. Recently, the effect of Lactoplantibacillus plantarum LS/07 (LB) was investigated in combination therapy with MTX in adjuvant arthritis (AA) in rats. LB administered in combination with MTX helped relieve hind leg swelling and body weight loss for the whole duration of the experiment. The combination therapy was more effective in reducing the inflammation than MTX alone. Regarding the positive anti-inflammatory effect of Lactiplantibacillus plantarum LS/07, administration of LB to AA rats reduced the concentrations of IL-17A, metalloproteinase-9 (MMP-9), and monocyte chemotactic protein-1 (MCP-1), with the latter two involved in macrophage recruitment and joint destruction during RA [91].

{kind=link}
Table 1.
The table reports the main studies on predisposing/protective gut bacteria involved in rheumatoid arthritis.
Table 1.
The table reports the main studies on predisposing/protective gut bacteria involved in rheumatoid arthritis.
| Metabolites | References | ||
|---|---|---|---|
| Predisposing Bacteria | Collinsella, Eggerthella, and Faecalibacterium genera | [32] | |
| Verrucomicrobiaand Akkermansia | 9,12-octadecadiynoic acid and 10Z-nonadecenoic acid | [33] (animal study) | |
| Lactobacillus salivarius | [62] | ||
| P. copri | [63] | ||
| Prevotella sp. | [64] | ||
| Protective Bacteria | B. fragilis | Butyrate | [52] (animal study) |
| Escherichia coli and Bacteroides species | [53] (animal study), [54] (animal study) | ||
| Haemophilus spp. | [62] | ||
| Lactiplantibacillus plantarumLS/07 | [82] (animal study) | ||
| P. distasonis | LCA, DCA, isoLCA and 3-oxoLCA | [86] (animal study) | |
| Bifidobacterium pseudocatenulatum | unconjugated secondary BA | [87] (animal study) | |
| L. casei | [88,89] (animal study), [90] (animal study) | ||
| L. acidophilus | [89] (animal study) |
LCA: lithocholic acid; DCA: deoxycholic acid; isoLCA: isolithocholic acid; 3-oxoLCA: 3-oxolithocholic acid; BA: bile acid.
5. Conclusions and Future Perspectives
The association between the gut microbiome and metabolites has not been fully evaluated yet in RA. Nevertheless, combined gut microbiome and metabolome studies have suggested promising new research directions to deepen our knowledge of RA pathogenesis.
The microbiome is critical for the balance of the host immune system, and its changes are essential for modulating inflammation and promoting the loss of immune tolerance, as demonstrated by the correlation between gut microbiota dysbiosis and the development of arthritis in several mouse models. The early stages of the pathological immune response associated with autoimmune diseases occur in mucosal tissues, such as the intestinal or oral mucosa, and are strongly associated with an abundance of specific bacterial species. Several studies, including animal studies, have pointed out an interaction between the gut microbiome, local/systemic immunity, and activation of joint inflammation. The link between these domains could be intimately connected to the activation of specific metabolic pathways. Metabolomics is an approach that has not been fully explored, but certainly holds promise for identifying biomarkers in different fields of medicine, including rheumatic diseases. Despite the undisputed potential, some limitations emerge, such as the lack of clear standardization of the technique used (NMR or MS) and sample collection methods, as well as the challenging availability of these methods and the specific laboratory skills required.
This review summarizes recent studies that have shown the correlation between the development of RA and the cross-link between the microbiome and metabolome.
Further evidence is needed to confirm what has been highlighted so far regarding the possible role of the microbiome and its influence on the metabolome in shaping the pathogenesis of RA. Identifying the prevalent metabolites and studying the gut–joint axis and intestinal dysbiosis could represent the fundamental steps needed to broaden the therapeutic strategies scenario in the era of personalized medicine to achieve a comprehensive “treat-to-target” approach. Moreover, the identification of these markers can be a valuable tool for predicting the course of the disease and preventing its progression.
Author Contributions
L.L.B. and C.R.: writing—original draft preparation, writing—review and editing, conceptualization; G.G. (Giulia Grasso) and F.M.: visualization, formal analysis, methodology, resources; F.C. (Federica Camarda): visualization, resources; F.C. (Francesco Ciccia): supervision, project administration; G.G. (Giuliana Guggino): supervision, project administration, conceptualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J.D. Cytokines in rheumatoid arthritis—Shaping the immunological landscape. Nat. Rev. Rheumatol. 2016, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Petrovská, N.; Prajzlerová, K.; Vencovský, J.; Šenolt, L.; Filková, M. The pre-clinical phase of rheumatoid arthritis: From risk factors to prevention of arthritis. Autoimmun. Rev. 2021, 20, 102797. [Google Scholar] [CrossRef] [PubMed]
- Romão, V.C.; Fonseca, J.E. Disease mechanisms in preclinical rheumatoid arthritis: A narrative review. Front. Med. 2022, 9, 689711. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in the development of rheumatoid arthritis. Immunol. Rev. 2020, 294, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed. J. 2021, 44, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Saferding, V.; Blüml, S. Innate immunity as the trigger of systemic autoimmune diseases. J. Autoimmun. 2020, 110, 102382. [Google Scholar] [CrossRef]
- Siouti, E.; Andreakos, E. The many facets of macrophages in rheumatoid arthritis. Biochem. Pharmacol. 2019, 165, 152–169. [Google Scholar] [CrossRef]
- Chan, K.-W.A.; Felson, D.T.; Yood, R.A.; Walker, A.M. The lag time between onset of symptoms and diagnosis of rheumatoid arthritis. Arthritis Rheum. 1994, 37, 814–820. [Google Scholar] [CrossRef]
- O’Dell, J.R. Treating rheumatoid arthritis early: A window of opportunity? Arthritis Rheum. 2002, 46, 283–285. [Google Scholar] [CrossRef]
- Xu, L.; Chang, C.; Jiang, P.; Wei, K.; Zhang, R.; Jin, Y.; Zhao, J.; Xu, L.; Shi, Y.; Guo, S.; et al. Metabolomics in rheumatoid arthritis: Advances and review. Front. Immunol. 2022, 13, 961708. [Google Scholar] [CrossRef] [PubMed]
- Coras, R.; Murillo-Saich, J.D.; Guma, M. Circulating Pro- and Anti-Inflammatory Metabolites and Its Potential Role in Rheumatoid Arthritis Pathogenesis. Cells 2020, 9, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chen, B.; Fang, Z.; Leng, Y.-F.; Wang, D.-W.; Chen, F.-Q.; Xu, X.; Sun, Z.-L. Metabolomics in the development and progression of rheumatoid arthritis: A systematic review. Jt. Bone Spine 2020, 87, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Camarda, F.; Donzella, D.; La Barbera, L.; Guggino, G. Metabolomics: An Emerging Approach to Understand Pathogenesis and to Assess Diagnosis and Response to Treatment in Spondyloarthritis. Cells 2022, 11, 549. [Google Scholar] [CrossRef]
- Jutley, G.S.; Sahota, K.; Sahbudin, I.; Filer, A.; Arayssi, T.; Young, S.P.; Raza, K. Relationship Between Inflammation and Metabolism in Patients With Newly Presenting Rheumatoid Arthritis. Front. Immunol. 2021, 12, 676105. [Google Scholar] [CrossRef]
- Young, S.P.; Kapoor, S.R.; Viant, M.R.; Byrne, J.J.; Filer, A.; Buckley, C.D.; Kitas, G.D.; Raza, K. The Impact of Inflammation on Metabolomic Profiles in Patients with Arthritis. Arthritis Rheum. 2013, 65, 2015–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietzner, M.; Kaul, A.; Henning, A.-K.; Kastenmüller, G.; Artati, A.; Lerch, M.M.; Adamski, J.; Nauck, M.; Friedrich, N. Comprehensive metabolic profiling of chronic low-grade inflammation among generally healthy individuals. BMC Med. 2017, 15, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, S.R.; Filer, A.; Fitzpatrick, M.A.; Fisher, B.A.; Taylor, P.C.; Buckley, C.D.; McInnes, I.B.; Raza, K.; Young, S.P. Metabolic Profiling Predicts Response to Anti–Tumor Necrosis Factor α Therapy in Patients With Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; for the IMID Consortium; Julià, A.; Vinaixa, M.; Domènech, E.; Fernández-Nebro, A.; Cañete, J.D.; Ferrándiz, C.; Tornero, J.; Gisbert, J.P. Urine metabolome profiling of immune-mediated inflammatory diseases. BMC Med. 2016, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Galvã¡n-Peã±A, S.; O’Neill, L.A.J. Metabolic Reprograming in Macrophage Polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4+ T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Zeisbrich, M.; Goronzy, J.J. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr. Opin. Immunol. 2017, 46, 112–120. [Google Scholar] [CrossRef]
- Kettunen, J.; Tukiainen, T.; Sarin, A.-P.; Ortega-Alonso, A.; Tikkanen, E.; Lyytikäinen, L.-P.; Kangas, A.J.; Soininen, P.; Würtz, P.; Silander, K.; et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat. Genet. 2012, 44, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Kettunen, J.; Demirkan, A.; Würtz, P.; Draisma, H.H.; Haller, T.; Rawal, R.; Vaarhorst, A.; Kangas, A.J.; Lyytikäinen, L.-P.; Pirinen, M.; et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun. 2016, 7, 11122. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Fernando, M.M.A.; Stevens, C.R.; Walsh, E.C.; De Jager, P.L.; Goyette, P.; Plenge, R.M.; Vyse, T.J.; Rioux, J.D. Defining the Role of the MHC in Autoimmunity: A Review and Pooled Analysis. PLOS Genet. 2008, 4, e1000024. [Google Scholar] [CrossRef]
- Rantapää-Dahlqvist, S.; de Jong, B.A.W.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- van Delft, M.A.; Huizinga, T.W. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ma, C.; Liu, L.; He, J.; Zhu, C.; Zheng, F.; Dai, W.; Hong, X.; Liu, D.; Tang, D.; et al. Analysis of gut microbiota and metabolites in patients with rheumatoid arthritis and identification of potential biomarkers. Aging 2021, 13, 23689–23701. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Wu, C.; Zeng, X.; Wang, Q. The role of gut microbiota in the pathogenesis of rheumatic diseases. Clin. Rheumatol. 2018, 37, 25–34. [Google Scholar] [CrossRef]
- Montgomery, A.B.; Kopec, J.; Shrestha, L.; Thezenas, M.-L.; Burgess-Brown, N.A.; Fischer, R.; Yue, W.W.; Venables, P.J. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: Implications for autoimmunity in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Janssen, K.M.; Vissink, A.; de Smit, M.J.; Westra, J.; Brouwer, E. Lessons to be learned from periodontitis. Curr. Opin. Rheumatol. 2013, 25, 241–247. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.-A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Marchesan, J.T.; Gerow, E.A.; Schaff, R.; Taut, A.D.; Shin, S.-Y.; Sugai, J.; Brand, D.; Burberry, A.; Jorns, J.; Lundy, S.K.; et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Res. Ther. 2013, 15, R186. [Google Scholar] [CrossRef] [Green Version]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans—Induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Beckler, M.D.; Amini, S.S.; Kesselman, M.M. Oral Microbiome in Pre-Rheumatoid Arthritis: The Role of Aggregatibacter Actinomycetemcomitans in Bacterial Composition. Cureus 2022, 5, e32201. [Google Scholar] [CrossRef]
- Looh, S.C.; Soo, Z.M.P.; Wong, J.J.; Yam, H.C.; Chow, S.K.; Hwang, J.S. Aggregatibacter actinomycetemcomitans as the Aetiological Cause of Rheumatoid Arthritis: What Are the Unsolved Puzzles? Toxins 2022, 14, 50. [Google Scholar] [CrossRef]
- Liu, X.; Zeng, B.; Zhang, J.; Li, W.; Mou, F.; Wang, H.; Zou, Q.; Zhong, B.; Wu, L.; Wei, H.; et al. Role of the Gut Microbiome in Modulating Arthritis Progression in Mice. Sci. Rep. 2016, 6, 30594. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Vaghef-Mehrabany, E.; Alipour, B.; Homayouni-Rad, A.; Sharif, S.-K.; Asghari-Jafarabadi, M.; Zavvari, S. Probiotic supplementation improves inflammatory status in patients with rheumatoid arthritis. Nutrition 2014, 30, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, J.; Hu, C.; Xie, Z.; Li, H.; Wei, S.; Wang, D.; Wen, C.; Xu, G. Exploration of the serum metabolite signature in patients with rheumatoid arthritis using gas chromatography–mass spectrometry. J. Pharm. Biomed. Anal. 2016, 127, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, T.; Maeda, Y.; Nii, T.; Motooka, D.; Matsumoto, Y.; Matsushita, M.; Matsuoka, H.; Yoshimura, M.; Kawada, S.; Teshigawara, S.; et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population. Ann. Rheum. Dis. 2020, 79, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Bahadori, B.; Uitz, E.; Thonhofer, R.; Trummer, M.; Pestemer-Lach, I.; McCarty, M.; Krejs, G.J. ω-3 Fatty Acids Infusions as Adjuvant Therapy in Rheumatoid Arthritis. J. Parenter. Enter. Nutr. 2010, 34, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häger, J.; Bang, H.; Hagen, M.; Frech, M.; Träger, P.; Sokolova, M.; Steffen, U.; Tascilar, K.; Sarter, K.; Schett, G.; et al. The Role of Dietary Fiber in Rheumatoid Arthritis Patients: A Feasibility Study. Nutrients 2019, 11, 2392. [Google Scholar] [CrossRef] [Green Version]
- La Barbera, L.; Macaluso, F.; Fasano, S.; Grasso, G.; Ciccia, F.; Guggino, G. Microbiome Changes in Connective Tissue Diseases and Vasculitis: Focus on Metabolism and Inflammation. Int. J. Mol. Sci. 2022, 23, 6532. [Google Scholar] [CrossRef]
- Luu, M.; Visekruna, A. Short-chain fatty acids: Bacterial messengers modulating the immunometabolism of T cells. Eur. J. Immunol. 2019, 49, 842–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Dong, C.; Zhao, B.; Lin, K.; Tian, Y.; Zhang, R.; Zhu, L.; Xu, H.; Yang, L. Bacteroides fragilis participates in the therapeutic effect of methotrexate on arthritis through metabolite regulation. Front. Microbiol. 2022, 13, 1015130. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, O.; Kohashi, Y.; Takahashi, T.; Ozawa, A.; Shigematsu, N. Reverse Effect of Gram-Positive Bacteria vs. Gram-Negative Bacteria on Adjuvant-Induced Arthritis in Germfree Rats. Microbiol. Immunol. 1985, 29, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Kohashi, O.; Kohashi, Y.; Takahashi, T.; Ozawa, A.; Shigematsu, N. Suppressive effect ofEscherichia coli on adjuvant-induced arthritis in germ-free rats. Arthritis Rheum. 1986, 29, 547–553. [Google Scholar] [CrossRef]
- Breban, M.A.; Moreau, M.C.; Fournier, C.; Ducluzeau, R.; Kahn, M.F. Influence of the bacterial flora on collagen-induced arthritis in susceptible and resistant strains of rats. Clin. Exp. Rheumatol. 1993, 11, 61–64. [Google Scholar] [PubMed]
- Sandberg, M.E.C.; Bengtsson, C.; Klareskog, L.; Alfredsson, L.; Saevarsdottir, S. Recent infections are associated with decreased risk of rheumatoid arthritis: A population-based case-control study. Ann. Rheum. Dis. 2015, 74, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.T.; Rooney, P.J.; Jones, D.B.; Bienenstock, J.; Goodacre, R.L. Increased intestinal permeability in patients with rheumatoid arthritis: A side-effect of oral nonsteroidal anti-inflammatory drug therapy? Rheumatology 1987, 26, 103–107. [Google Scholar] [CrossRef]
- Segal, A.; Isenberg, D.; Hajirousou, V.; Tolfree, S.; Clark, J.; Snaith, M.L. Preliminary evidence for gut involvement in the pathogenesis of rheumatoid arthritis? Rheumatology 1986, 25, 162–166. [Google Scholar] [CrossRef]
- Macaluso, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Ciccia, F. Histopathology of the gut in rheumatic diseases. Reumatismo 2018, 70, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Porzio, V.; Biasi, G.; Corrado, A.; De Santi, M.; Vindigni, C.; Vrti, S.; Bayeli, P.F.; Marcolongo, R. Intestinal Histological and Ultrastructural Inflammatory Changes in Spondyloarthropathy and Rheumatoid Arthritis. Scand. J. Rheumatol. 1997, 26, 92–98. [Google Scholar] [CrossRef]
- Mielants, H.; De Vos, M.; Goemaere, S.; Schelstraete, K.; Cuvelier, C.; Goethals, K.; Maertens, M.; Ackerman, C.; Veys, E.M. Intestinal mucosal permeability in inflammatory rheumatic diseases. II. Role of disease. J. Rheumatol. 1991, 18, 394–400. [Google Scholar] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Pianta, A.; Arvikar, S.L.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.; Steere, A.C. Two rheumatoid arthritis–specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Investig. 2017, 127, 2946–2956. [Google Scholar] [CrossRef]
- Radu, A.-F.; Bungau, S.G.; Negru, P.A.; Marcu, M.F.; Andronie-Cioara, F.L. In-depth bibliometric analysis and current scientific mapping research in the context of rheumatoid arthritis pharmacotherapy. Biomed. Pharmacother. 2022, 154, 113614. [Google Scholar] [CrossRef] [PubMed]
- Bartikoski, B.J.; De Oliveira, M.S.; Santo, R.C.D.E.; Dos Santos, L.P.; Dos Santos, N.G.; Xavier, R.M. A Review of Metabolomic Profiling in Rheumatoid Arthritis: Bringing New Insights in Disease Pathogenesis, Treatment and Comorbidities. Metabolites 2022, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Plant, D.; Verstappen, S.M.; Isaacs, J.D.; Morgan, A.; Hyrich, K.L.; Barton, A.; Wilson, A.G. The MATURA investigators Differential DNA methylation correlates with response to methotrexate in rheumatoid arthritis. Rheumatology 2020, 59, 1364–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbaniak, B.; Plewa, S.; Klupczynska, A.; Sikorska, D.; Samborski, W.; Kokot, Z.J. Serum free amino acid levels in rheumatoid arthritis according to therapy and physical disability. Cytokine 2019, 113, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Priori, R.; Scrivo, R.; Brandt, J.; Valerio, M.; Casadei, L.; Valesini, G.; Manetti, C. Metabolomics in rheumatic diseases: The potential of an emerging methodology for improved patient diagnosis, prognosis, and treatment efficacy. Autoimmun. Rev. 2013, 12, 1022–1030. [Google Scholar] [CrossRef]
- Ramalho, R.; Rao, M.; Zhang, C.; Agrati, C.; Ippolito, G.; Wang, F.-S.; Zumla, A.; Maeurer, M. Immunometabolism: New insights and lessons from antigen-directed cellular immune responses. Semin. Immunopathol. 2020, 42, 279–313. [Google Scholar] [CrossRef]
- Gosselt, H.R.; Muller, I.B.; Jansen, G.; Van Weeghel, M.; Vaz, F.M.; Hazes, J.M.W.; Heil, S.G.; De Jonge, R. Identification of Metabolic Biomarkers in Relation to Methotrexate Response in Early Rheumatoid Arthritis. J. Pers. Med. 2020, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Salamoun, Y.M.; Polireddy, K.; Cho, Y.K.; Medcalf, M.R.; Funk, R.S. Methotrexate Disposition, Anti-Folate Activity, and Metabolomic Profiling to Identify Molecular Markers of Disease Activity and Drug Response in the Collagen-Induced Arthritis Mouse Model. Metabolites 2021, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Huang, J.; Fan, H.; He, D.; Zhao, S.; Shu, Y.; Li, H.; Liu, L.; Lu, S.; Xiao, C.; et al. Treatment of Rheumatoid Arthritis Using Combination of Methotrexate and Tripterygium Glycosides Tablets—A Quantitative Plasma Pharmacochemical and Pseudotargeted Metabolomic Approach. Front. Pharmacol. 2018, 9, 1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artacho, A.; Isaac, S.; Nayak, R.; Flor-Duro, A.; Alexander, M.; Koo, I.; Manasson, J.; Smith, P.B.; Rosenthal, P.; Homsi, Y.; et al. The Pretreatment Gut Microbiome Is Associated With Lack of Response to Methotrexate in New-Onset Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and Abnormal Glucose and Lipid Metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priori, R.; Casadei, L.; Valerio, M.; Scrivo, R.; Valesini, G.; Manetti, C. 1H-NMR-Based Metabolomic Study for Identifying Serum Profiles Associated with the Response to Etanercept in Patients with Rheumatoid Arthritis. PLoS ONE 2015, 10, e0138537. [Google Scholar] [CrossRef]
- Tatar, Z.; Migne, C.; Petera, M.; Gaudin, P.; Lequerre, T.; Marotte, H.; Tebib, J.; Guillot, E.P.; Soubrier, M. Variations in the metabolome in response to disease activity of rheumatoid arthritis. BMC Musculoskelet. Disord. 2016, 17, 353. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Saegusa, J.; Onishi, A.; Morinobu, A. Biomarkers identified by serum metabolomic analysis to predict biologic treatment response in rheumatoid arthritis patients. Rheumatology 2019, 58, 2153–2161. [Google Scholar] [CrossRef]
- Teitsma, X.M.; Yang, W.; Jacobs, J.W.G.; Pethö-Schramm, A.; Borm, M.E.A.; Harms, A.C.; Hankemeier, T.; van Laar, J.M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Baseline metabolic profiles of early rheumatoid arthritis patients achieving sustained drug-free remission after initiating treat-to-target tocilizumab, methotrexate, or the combination: Insights from systems biology. Arthritis Res. Ther. 2018, 20, 230. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Saich, J.D.; Diaz-Torne, C.; Ortiz, M.A.; Coras, R.; Gil-Alabarse, P.; Pedersen, A.; Corominas, H.; Vidal, S.; Guma, M. Metabolomics profiling predicts outcome of tocilizumab in rheumatoid arthritis: An exploratory study. Metabolomics 2021, 17, 74. [Google Scholar] [CrossRef]
- Sweeney, S.R.; Kavanaugh, A.; Lodi, A.; Wang, B.; Boyle, D.; Tiziani, S.; Guma, M. Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open 2016, 2, e000289. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Naved, T.; Bhatia, S.; Al-Harrasi, A.; Chakrabarti, P.; Aleya, L.; et al. Mechanistic insights into the role of B cells in rheumatoid arthritis. Int. Immunopharmacol. 2021, 99, 108078. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Chen, P.-K.; Chen, C.-C.; Chang, S.-H.; Chen, C.-H.; Chen, D.-Y. Increased Levels of Omega-3 Fatty Acids and DHA Are Linked to Pain Reduction in Rheumatoid Arthritis Patients Treated with Janus Kinase Inhibitors. Nutrients 2021, 13, 3050. [Google Scholar] [CrossRef]
- Cuppen, B.V.J.; Fu, J.; van Wietmarschen, H.A.; Harms, A.C.; Koval, S.; Marijnissen, A.C.A.; Peeters, J.J.W.; Bijlsma, J.W.J.; Tekstra, J.; van Laar, J.M.; et al. Exploring the Inflammatory Metabolomic Profile to Predict Response to TNF-α Inhibitors in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0163087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani, B.; Golkar, H.R.; Farshbaf, S.; Emadi-Baygi, M.; Tajabadi-Ebrahimi, M.; Jafari, P.; Akhavan, R.; Taghizadeh, M.; Memarzadeh, M.R.; Asemi, Z. Clinical and metabolic response to probiotic supplementation in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled trial. Int. J. Rheum. Dis. 2016, 19, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Guo, Y.; Wang, H.; Yin, A.; Hu, J.; Yuan, T.; Zhou, S.; Xu, W.; Wei, P.; Yin, S.; et al. Gut commensal Parabacteroides distasonis alleviates inflammatory arthritis. Gut 2023. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, H.; Yang, N.; Xia, X.; Chen, Q.; Zhou, D.; Liu, Z.; Chen, X.; Chen, Y.; Huang, W.; et al. Bifidobacterium pseudocatenulatum-Mediated Bile Acid Metabolism to Prevent Rheumatoid Arthritis via the Gut–Joint Axis. Nutrients 2023, 15, 255. [Google Scholar] [CrossRef] [PubMed]
- Alipour, B.; Homayouni-Rad, A.; Vaghef-Mehrabany, E.; Sharif, S.K.; Vaghef-Mehrabany, L.; Asghari-Jafarabadi, M.; Nakhjavani, M.R.; Mohtadi-Nia, J. Effects of Lactobacillus casei supplementation on disease activity and inflammatory cytokines in rheumatoid arthritis patients: A randomized double-blind clinical trial. Int. J. Rheum. Dis. 2014, 17, 519–527. [Google Scholar] [CrossRef]
- Amdekar, S.; Singh, V.; Kumari, A.; Sharma, P.; Singh, R.; Vaghef-Mehrabany, E.; Homayouni-Rad, A.; Alipour, B.; Sharif, S.-K.; Vaghef-Mehrabany, L.; et al. Lactobacillus casei and Lactobacillus acidophilus Regulate Inflammatory Pathway and Improve Antioxidant Status in Collagen-Induced Arthritic Rats. J. Interf. Cytokine Res. 2013, 33, 1–8. [Google Scholar] [CrossRef]
- Amdekar, S.; Singh, V.; Singh, R.; Sharma, P.; Keshav, P.; Kumar, A. Lactobacillus casei reduces the Inflammatory Joint Damage Associated with Collagen-Induced Arthritis (CIA) by Reducing the Pro-Inflammatory Cytokines: Lactobacillus Casei: COX-2 Inhibitor. J. Clin. Immunol. 2011, 31, 147–154. [Google Scholar] [CrossRef]
- Pružinská, K.; Slovák, L.; Dráfi, F.; Poništ, S.; Juránek, I.; Chrastina, M.; Švík, K.; Strojný, L.; Ambro, L.; Bauerová, K. Enhanced Anti-Inflammatory Effect of the Combination of Lactiplantibacillus plantarum LS/07 with Methotrexate Compared to Their Monotherapies Studied in Experimental Arthritis. Molecules 2022, 28, 297. [Google Scholar] [CrossRef] [PubMed]
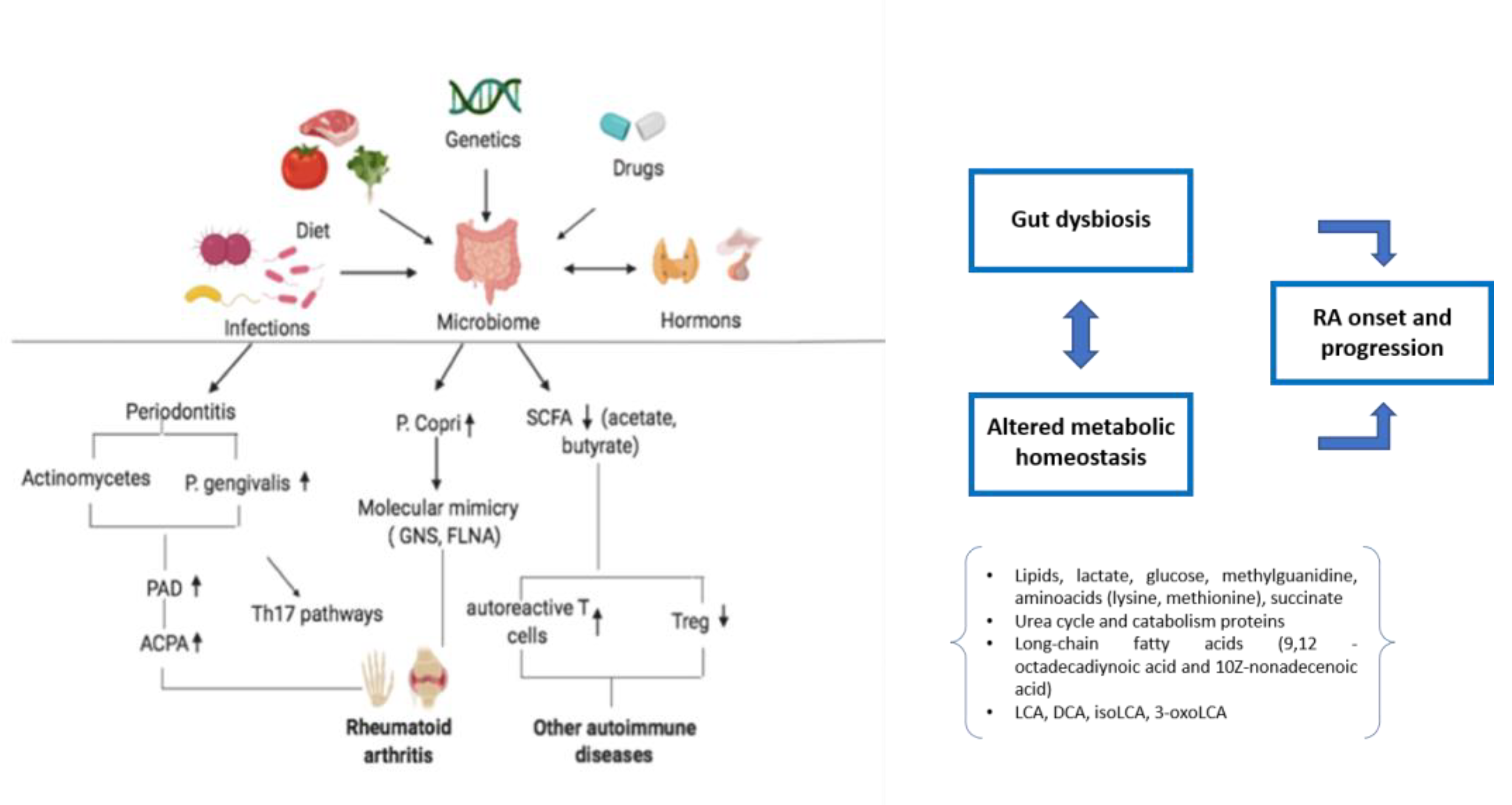
Figure 1.
Dysbiosis and altered metabolic homeostasis in rheumatoid arthritis. Genetics, infections, and other environmental factors are involved in dysbiosis, triggering the immunological mechanisms underlying RA development. Gut dysbiosis and altered metabolic pathways contribute to RA onset and progression. RA: rheumatoid arthritis; PAD: peptidyl-arginine deiminases; ACPA: anti-citrullinated protein antibodies; SCFA: short-chain fatty acids; LCA: lithocholic acid; DCA: deoxycholic acid; isoLCA: isolithocholic acid; 3-oxoLCA: 3-oxolithocholic acid.
Figure 1.
Dysbiosis and altered metabolic homeostasis in rheumatoid arthritis. Genetics, infections, and other environmental factors are involved in dysbiosis, triggering the immunological mechanisms underlying RA development. Gut dysbiosis and altered metabolic pathways contribute to RA onset and progression. RA: rheumatoid arthritis; PAD: peptidyl-arginine deiminases; ACPA: anti-citrullinated protein antibodies; SCFA: short-chain fatty acids; LCA: lithocholic acid; DCA: deoxycholic acid; isoLCA: isolithocholic acid; 3-oxoLCA: 3-oxolithocholic acid.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
La Barbera, L.; Rizzo, C.; Grasso, G.; Macaluso, F.; Camarda, F.; Ciccia, F.; Guggino, G. The Cross-Talk between Microbiome and Metabolome in Rheumatoid Arthritis. BioChem 2023, 3, 47-60. https://doi.org/10.3390/biochem3010004
AMA Style
La Barbera L, Rizzo C, Grasso G, Macaluso F, Camarda F, Ciccia F, Guggino G. The Cross-Talk between Microbiome and Metabolome in Rheumatoid Arthritis. BioChem. 2023; 3(1):47-60. https://doi.org/10.3390/biochem3010004
Chicago/Turabian StyleLa Barbera, Lidia, Chiara Rizzo, Giulia Grasso, Federica Macaluso, Federica Camarda, Francesco Ciccia, and Giuliana Guggino. 2023. "The Cross-Talk between Microbiome and Metabolome in Rheumatoid Arthritis" BioChem 3, no. 1: 47-60. https://doi.org/10.3390/biochem3010004