
Exploring the Interplay between Fatty Acids, Inflammation, and Type 2 Diabetes

 ,
,  , and
, and
Abstract
:1. Introduction
2. Diet, Adipose Tissue, and Type 2 Diabetes
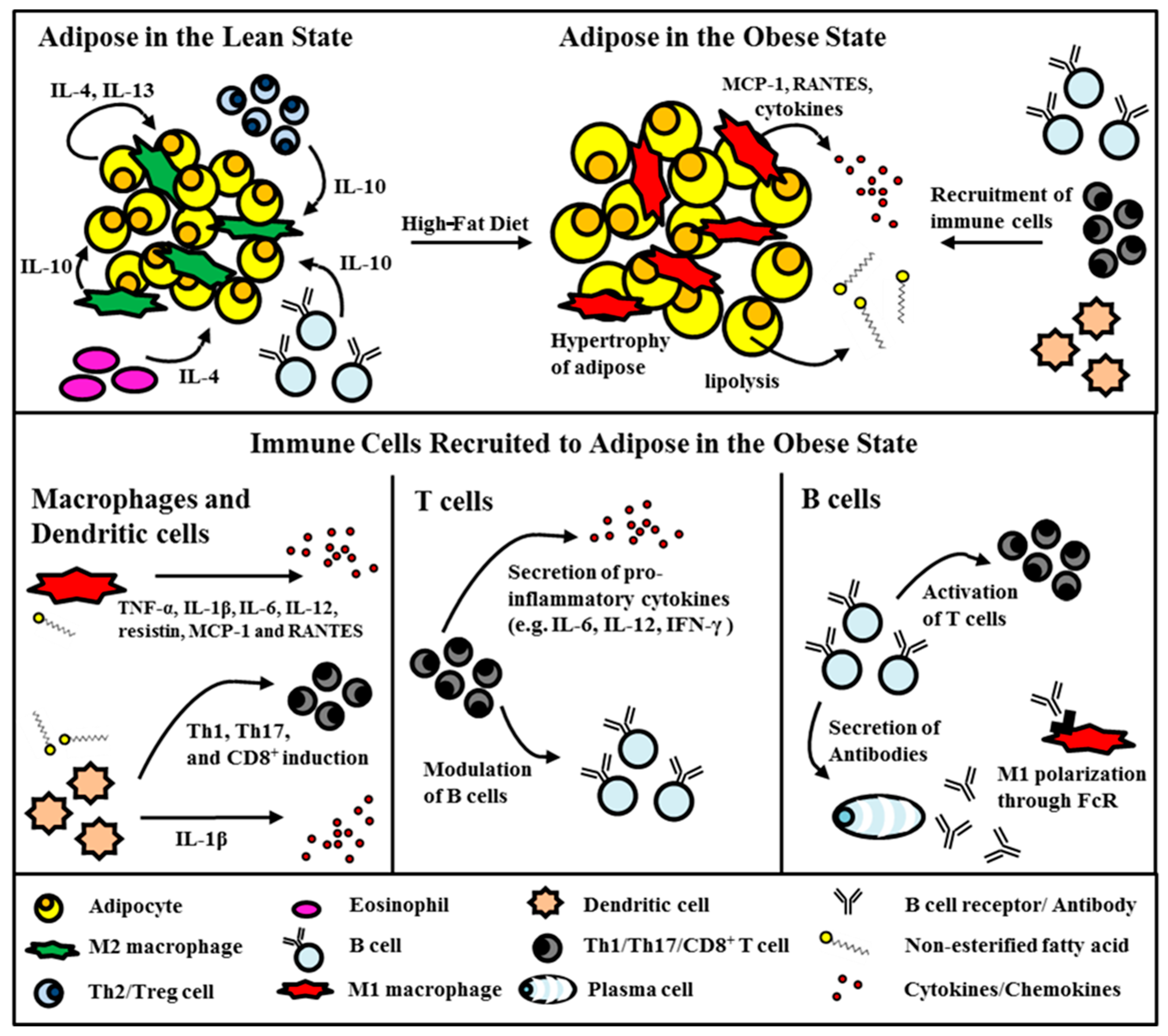
2.1. Adipose Tissue in the Obese State
2.2. The Role of Fatty Acids in Triggering Insulin-Secreting Beta-Cells’ Failure
2.3. Dietary Fatty Acids and Type 2 Diabetes
Various potential pathways have been suggested for the advancement of clinical treatments utilizing FFA receptor agonism. Among these is the exploration of co-therapeutic strategies combining FFA receptor agonists with existing type 2 diabetes mellitus (T2DM) therapies. For instance, AS2575959, functioning as an FFA1 agonist, demonstrates synergistic effects when used alongside a DPP-IV inhibitor, leading to enhanced glucose homeostasis [41]. DS-1558, functioning as an FFA1 agonist, exhibits synergistic effects when combined with exendin-4, leading to improved glucose homeostasis in diabetic mice [42].
2.4. Saturated Fatty Acid Stimulation of Immune Cells
3. Visceral Fat Inflammation in Type 2 Diabetes
3.1. Monocytes/Macrophages
3.2. Dendritic Cells
3.3. T Cells
3.4. B Cells
3.5. Other Immune Cells
4. Systemic Inflammation in Type 2 Diabetes
5. Novel Concepts in Diet-Induced Inflammation and Type 2 Diabetes
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, F.B. Globalization of diabetes: The role of diet, lifestyle, and genes. Diabetes Care 2011, 34, 1249–1257. [Google Scholar] [CrossRef]
- Franks, P.W.; Hanson, R.L.; Knowler, W.C.; Sievers, M.L.; Bennett, P.H.; Looker, H.C. Childhood obesity, other cardiovascular risk factors, and premature death. N. Engl. J. Med. 2010, 362, 485–493. [Google Scholar] [CrossRef]
- American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013, 36, 1033–1046. [Google Scholar] [CrossRef]
- Wheeler, G.; Montgomery, S.B.; Beeson, L.; Bahjri, K.; Shulz, E.; Firek, A.; De Leon, M.; Cordero-MacIntyre, Z. En Balance: The effects of Spanish diabetes education on physical activity changes and diabetes control. Diabetes Educ. 2012, 38, 723–732. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Winer, S.; Winer, D.A. The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol. Cell Biol. 2012, 90, 755–762. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Minze, L.J.; Lin, J.; Zou, J.; Liu, J.Z.; Ren, Y.; Yin, Z.; Hamilton, D.J.; Reardon, P.R.; et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 2013, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Jager, J.; Paulmyer-Lacroix, O.; Peiretti, F.; Gremeaux, T.; Verdier, M.; Grino, M.; Stepanian, A.; Msika, S.; Burcelin, R.; et al. The inflammatory receptor CD40 is expressed on human adipocytes: Contribution to crosstalk between lymphocytes and adipocytes. Diabetologia 2009, 52, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Gram, H.; Zorrilla, E.P.; Conti, B.; Bartfai, T. Insights into the roles of the inflammatory mediators IL-1, IL-18 and PGE2 in obesity and insulin resistance. Swiss Med. Wkly. 2008, 138, 665–673. [Google Scholar] [CrossRef]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ghosh, S.; Perrard, X.D.; Feng, L.; Garcia, G.E.; Perrard, J.L.; Sweeney, J.F.; Peterson, L.E.; Chan, L.; Smith, C.W.; et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007, 115, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lair, B.; Laurens, C.; Van Den Bosch, B.; Moro, C. Novel Insights and Mechanisms of Lipotoxicity-Driven Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6358. [Google Scholar] [CrossRef] [PubMed]
- Véret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbé, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Higa, M.; Zhou, Y.T.; Wang, M.Y.; Newgard, C.B.; Unger, R.H. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 1998, 273, 32487–32490. [Google Scholar]
- Miyauchi, S.; Hirasawa, A.; Ichimura, A.; Hara, T.; Tsujimoto, G. New frontiers in gut nutrient sensor research: Free fatty acid sensing in the gastrointestinal tract. J. Pharmacol. Sci. 2010, 112, 19–24. [Google Scholar] [CrossRef]
- Steneberg, P.; Rubins, N.; Bartoov-Shifman, R.; Walker, M.D.; Edlund, H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005, 1, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [PubMed]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Perret, V.; Favre, D.; Abderrahmani, A.; Yang, J.Y.; Widmann, C.; Regazzi, R. Role of the transcriptional factor C/EBPbeta in free fatty acid-elicited beta-cell failure. Mol. Cell. Endocrinol. 2009, 305, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.S.; Karin, M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef]
- Storlien, L.H.; Jenkins, A.B.; Chisholm, D.J.; Pascoe, W.S.; Khouri, S.; Kraegen, E.W. Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 1991, 40, 280–289. [Google Scholar] [CrossRef]
- Oliver, E.; McGillicuddy, F.; Phillips, C.; Toomey, S.; Roche, H.M. The role of inflammation and macrophage accumulation in the development of obesity-induced type 2 diabetes mellitus and the possible therapeutic effects of long-chain n-3 PUFA. Proc. Nutr. Soc. 2010, 69, 232–243. [Google Scholar] [CrossRef]
- Kolb, H.; Mandrup-Poulsen, T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia 2010, 53, 10–20. [Google Scholar] [CrossRef]
- Riserus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef]
- Salgin, B.; Ong, K.K.; Thankamony, A.; Emmett, P.; Wareham, N.J.; Dunger, D.B. Higher fasting plasma free fatty acid levels are associated with lower insulin secretion in children and adults and a higher incidence of type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3302–3309. [Google Scholar] [CrossRef]
- Paolisso, G.; Tataranni, P.A.; Foley, J.E.; Bogardus, C.; Howard, B.V.; Ravussin, E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia 1995, 38, 1213–1217. [Google Scholar] [CrossRef]
- Charles, M.A.; Eschwege, E.; Thibult, N.; Claude, J.-R.; Warnet, J.-M.; Rosselin, G.E.; Girard, J.; Balkau, B. The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: Results of the Paris Prospective Study. Diabetologia 1997, 40, 1101–1106. [Google Scholar] [CrossRef]
- Pankow, J.S.; Duncan, B.B.; Schmidt, M.I.; Ballantyne, C.M.; Couper, D.J.; Hoogeveen, R.C.; Golden, S.H. Fasting plasma free fatty acids and risk of type 2 diabetes: The atherosclerosis risk in communities study. Diabetes Care 2004, 27, 77–82. [Google Scholar] [CrossRef]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; DeFronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef]
- Boden, G.; Cheung, P.; Stein, T.P.; Kresge, K.; Mozzoli, M. FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E12–E19. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X.; Rosner, J.; Barton, M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes 1995, 44, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aljada, A.; Dandona, P. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Tantiwong, P.; Sriwijitkamol, A.; Shanmugasundaram, K.; Mohan, S.; Espinoza, S.; DeFronzo, R.A.; Dubé, J.J.; Musi, N. Effect of a sustained reduction in plasma free fatty acid concentration on insulin signalling and inflammation in skeletal muscle from human subjects. J. Physiol. 2013, 591, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yoshida, S.; Minoura, H.; Negoro, K.; Shimaya, A.; Shimokawa, T.; Shibasaki, M. Novel GPR40 agonist AS2575959 exhibits glucose metabolism improvement and synergistic effect with sitagliptin on insulin and incretin secretion. Life Sci. 2014, 94, 115–121. [Google Scholar] [CrossRef]
- Nakashima, R.; Yano, T.; Ogawa, J.; Tanaka, N.; Toda, N.; Yoshida, M.; Takano, R.; Inoue, M.; Honda, T.; Kume, S.; et al. Potentiation of insulin secretion and improvement of glucose intolerance by combining a novel G protein-coupled receptor 40 agonist DS-1558 with glucagon-like peptide-1 receptor agonists. Eur. J. Pharmacol. 2014, 737, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Yuan, X.; Shimoda, Y.; Uchio-Yamada, K.; Nakagawa, N.; Shirakawa, I.; Usami, T.; Tsukahara, T.; Nakayama, K.; Miyamoto, Y.; et al. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circ. Res. 2009, 105, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.-K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef]
- Davis, J.E.; Gabler, N.K.; Walker-Daniels, J.; Spurlock, M.E. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity 2008, 16, 1248–1255. [Google Scholar] [CrossRef]
- Suganami, T.; Mieda, T.; Itoh, M.; Shimoda, Y.; Kamei, Y.; Ogawa, Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem. Biophys. Res. Commun. 2007, 354, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Saberi, M.; Woods, N.-B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.E.; Weisberg, S.; Vardhana, P.; Tortoriello, D.V. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int. J. Obes. 2008, 32, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Wueest, S.; Rapold, R.A.; Schumann, D.M.; Rytka, J.M.; Schildknecht, A.; Nov, O.; Chervonsky, A.V.; Rudich, A.; Schoenle, E.J.; Donath, M.Y.; et al. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J. Clin. Investig. 2010, 120, 191–202. [Google Scholar] [CrossRef]
- Patsouris, D.; Li, P.P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef]
- Mbongue, J.; Nicholas, D.; Firek, A.; Langridge, W. The role of dendritic cells in tissue-specific autoimmunity. J. Immunol. Res. 2014, 2014, 857143. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Hashimoto, D.; Miller, J.; Merad, M. Dendritic cell and macrophage heterogeneity in vivo. Immunity 2011, 35, 323–335. [Google Scholar] [CrossRef]
- Stefanovic-Racic, M.; Yang, X.; Turner, M.S.; Mantell, B.S.; Stolz, D.B.; Sumpter, T.L.; Sipula, I.J.; Dedousis, N.; Scott, D.K.; Morel, P.A.; et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes 2012, 61, 2330–2339. [Google Scholar] [CrossRef]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef]
- Jaensson, E.; Uronen-Hansson, H.; Pabst, O.; Eksteen, B.; Tian, J.; Coombes, J.L.; Berg, P.L.; Davidsson, T.; Powrie, F.; Johansson-Lindbom, B.; et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008, 205, 2139–2149. [Google Scholar] [CrossRef]
- Nicholas, D.A.; Zhang, K.; Hung, C.; Glasgow, S.; Aruni, A.W.; Unternaehrer, J.; Payne, K.J.; Langridge, W.H.R.; De Leon, M. Palmitic acid is a toll-like receptor 4 ligand that induces human dendritic cell secretion of IL-1β. PLoS ONE 2017, 12, e0176793. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Abu-Asab, M.; Glasow, A.; Päth, G.; Hauner, H.; Tsokos, M.; Chrousos, G.P.; Scherbaum, W.A. Immunohistochemical and ultrastructural localization of leptin and leptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes 2000, 49, 532–538. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Zeyda, M.; Huber, J.; Prager, G.; Stulnig, T.M. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity 2011, 19, 743–748. [Google Scholar] [CrossRef]
- Kendall, M.R.; Hupfeld, C.J. FTY720, a sphingosine-1-phosphate receptor modulator, reverses high-fat diet-induced weight gain, insulin resistance and adipose tissue inflammation in C57BL/6 mice. Diabetes Obes. Metab. 2008, 10, 802–805. [Google Scholar] [CrossRef]
- Strissel, K.J.; DeFuria, J.; Shaul, M.E.; Bennett, G.; Greenberg, A.S.; Obin, M.S. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity 2010, 18, 1918–1925. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef]
- O’rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar] [CrossRef]
- Hong, C.-P.; Park, A.; Yang, B.-G.; Yun, C.H.; Kwak, M.-J.; Lee, G.-W.; Kim, J.-H.; Jang, M.S.; Lee, E.-J.; Jeun, E.-J.; et al. Gut-Specific Delivery of T-Helper 17 Cells Reduces Obesity and Insulin Resistance in Mice. Gastroenterology 2017, 152, 1998–2010. [Google Scholar] [CrossRef]
- Endo, Y.; Yokote, K.; Nakayama, T. The obesity-related pathology and Th17 cells. Cell. Mol. Life Sci. 2017, 74, 1231–1245. [Google Scholar] [CrossRef]
- Ip, B.; Cilfone, N.A.; Belkina, A.C.; DeFuria, J.; Jagannathan-Bogdan, M.; Zhu, M.; Kuchibhatla, R.; McDonnell, M.E.; Xiao, Q.; Kepler, T.B.; et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity 2016, 24, 102–112. [Google Scholar] [CrossRef]
- Vega-Cárdenas, M.; Uresti-Rivera, E.E.; Cortés-García, J.D.; Briones-Espinoza, M.; Ruíz-Rodríguez, V.M.; Reynaga-Hernández, E.; Mendez-Mancilla, A.; Portales-Pérez, D.P. Increased levels of adipose tissue-resident Th17 cells in obesity associated with miR-326. Immunol. Lett. 2019, 211, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, L.A.; Shen, W.-J.; Joyce-Shaikh, B.; Pyatnova, E.A.; Richards, A.G.; Thom, C.; Andrade, S.M.; Cua, D.J.; Kraemer, F.B.; Butcher, E.C. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J. Immunol. 2010, 185, 6947–6959. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Mogi, M.; Jing, F.; Iwanami, J.; Tsukuda, K.; Min, L.-J.; Higaki, J.; Horiuchi, M. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension 2012, 59, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Griffin, D.O.; Holodick, N.E.; Quach, T.D.; Kaku, H. Human B-1 cells take the stage. Ann. N. Y. Acad. Sci. 2013, 1285, 97–114. [Google Scholar] [CrossRef]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; DiLillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Wu, L.; Parekh, V.V.; Hsiao, J.; Kitamura, D.; Van Kaer, L. Spleen supports a pool of innate-like B cells in white adipose tissue that protects against obesity-associated insulin resistance. Proc. Natl. Acad. Sci. USA 2014, 111, E4638–E4647. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Takaki, S.; Nagasaki, M.; Otsu, M.; Yamashita, H.; Sugita, J.; Yoshimura, K.; Eto, K.; Komuro, I.; et al. Adipose Natural Regulatory B Cells Negatively Control Adipose Tissue Inflammation. Cell Metab. 2013, 18, 759–766. [Google Scholar] [CrossRef]
- Shen, L.; Yen Chng, M.H.; Alonso, M.N.; Yuan, R.; Winer, D.A.; Engleman, E.G. B-1a lymphocytes attenuate insulin resistance. Diabetes 2014, 64, 593–603. [Google Scholar] [CrossRef]
- Duffaut, C.; Galitzky, J.; Lafontan, M.; Bouloumie, A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem. Biophys. Res. Commun. 2009, 384, 482–485. [Google Scholar] [CrossRef]
- Jagannathan, M.; McDonnell, M.; Liang, Y.; Hasturk, H.; Hetzel, J.; Rubin, D.; Kantarci, A.; Van Dyke, T.E.; Ganley-Leal, L.M.; Nikolajczyk, B.S. Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia 2010, 53, 1461–1471. [Google Scholar] [CrossRef]
- Jagannathan, M.; McDonnell, M.; Liang, Y.; Hasturk, H.; Hetzel, J.; Rubin, D.; Kantarci, A.; Van Dyke, T.E.; Ganley-Leal, L.M.; Nikolajczyk, B.S. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar]
- Elgazar-Carmon, V.; Rudich, A.; Hadad, N.; Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef]
- Wu, D.; Molofsky, A.B.; Liang, H.-E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clément, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef]
- Ohmura, K.; Ishimori, N.; Ohmura, Y.; Tokuhara, S.; Nozawa, A.; Horii, S.; Andoh, Y.; Fujii, S.; Iwabuchi, K.; Onoé, K.; et al. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Mantell, B.S.; Stefanovic-Racic, M.; Yang, X.; Dedousis, N.; Sipula, I.J.; O’Doherty, R.M. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS ONE 2011, 6, e19831. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Sun, J.; Huang, J.; He, Y.; Yu, A.; Yu, X.; Yang, Z. TNF-alpha reduces g0s2 expression and stimulates lipolysis through PPAR-gamma inhibition in 3T3-L1 adipocytes. Cytokine 2014, 69, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Tillison, K.; Lee, J.H.; Rearick, D.A.; Smas, C.M. The adipose tissue triglyceride lipase ATGL/PNPLA2 is downregulated by insulin and TNF-alpha in 3T3-L1 adipocytes and is a target for transactivation by PPARgamma. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E115–E127. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Heckmann, B.L.; Lu, X.; Liu, J. Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-alpha (TNF-alpha)-induced lipolysis in adipocytes. J. Biol. Chem. 2011, 286, 40477–40485. [Google Scholar] [CrossRef] [PubMed]
- Stagakis, I.; Bertsias, G.; Karvounaris, S.; Kavousanaki, M.; Virla, D.; Raptopoulou, A.; Kardassis, D.; Boumpas, D.T.; Sidiropoulos, P.I. Anti-tumor necrosis factor therapy improves insulin resistance, beta cell function and insulin signaling in active rheumatoid arthritis patients with high insulin resistance. Arthritis Res. Ther. 2012, 14, R141. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C.; Vazquez-Rodriguez, T.R.; Miranda-Filloy, J.A.; Llorca, J. Insulin resistance in rheumatoid arthritis: The impact of the anti-TNF-alpha therapy. Ann. N. Y. Acad. Sci. 2010, 1193, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ferraz-Amaro, I.; Arce-Franco, M.; Muñiz, J.; López-Fernández, J.; Hernández-Hernández, V.; Franco, A.; Quevedo, J.; Martínez-Martín, J.; Díaz-González, F. Systemic blockade of TNF-alpha does not improve insulin resistance in humans. Horm. Metab. Res. = Horm.-Und Stoffwechselforschung = Horm. Metab. 2011, 43, 801–808. [Google Scholar]
- Bernstein, L.E.; Berry, J.; Kim, S.; Canavan, B.; Grinspoon, S.K. Effects of etanercept in patients with the metabolic syndrome. Arch. Intern. Med. 2006, 166, 902–908. [Google Scholar] [CrossRef]
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J. Biol. Chem. 2009, 284, 31936–31944. [Google Scholar] [CrossRef]
- Nathan, C.F.; Murray, H.W.; Wiebe, M.E.; Rubin, B.Y. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 1983, 158, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef]
- Winer, S.; Paltser, G.; Chan, Y.; Tsui, H.; Engleman, E.; Winer, D.; Dosch, H.M. Obesity predisposes to Th17 bias. Eur. J. Immunol. 2009, 39, 2629–2635. [Google Scholar] [CrossRef]
- Ogata, A.; Morishima, A.; Hirano, T.; Hishitani, Y.; Hagihara, K.; Shima, Y.; Narazaki, M.; Tanaka, T. Improvement of HbA1c during treatment with humanised anti-interleukin 6 receptor antibody, tocilizumab. Ann. Rheum. Dis. 2011, 70, 1164–1165. [Google Scholar] [CrossRef]
- Legrand-Poels, S.; Esser, N.; L’Homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Ehses, J.A.; Donath, M.Y.; Mandrup-Poulsen, T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 2009, 32, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Ojo, E.; Beeson, L.; Shulz, E.; Firek, A.; De Leon, M.; Balcazar, H.; Cordero-Macintyre, Z. Effect of the EnBalance, a culturally and language-sensitive diabetes education program, on dietary changes and plasma lipid profile in Hispanic diabetics. Int. J. Body Compos. Res. 2010, 8, S69–S76. [Google Scholar] [PubMed]
- Salto, L.M.; Cordero-MacIntyre, Z.; Beeson, L.; Schulz, E.; Firek, A.; De Leon, M. En Balance participants decrease dietary fat and cholesterol intake as part of a culturally sensitive Hispanic diabetes education program. Diabetes Educ. 2011, 37, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Palming, J.; Gabrielsson, B.G.; Jennische, E.; Smith, U.; Carlsson, B.; Carlsson, L.M.; Lönn, M. Plasma cells and Fc receptors in human adipose tissue--lipogenic and anti-inflammatory effects of immunoglobulins on adipocytes. Biochem. Biophys. Res. Commun. 2006, 343, 43–48. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Jagannathan-Bogdan, M.; McDonnell, M.E.; Shin, H.; Rehman, Q.; Hasturk, H.; Apovian, C.M.; Nikolajczyk, B.S. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J. Immunol. 2011, 186, 1162–1172. [Google Scholar] [CrossRef]
- Jager, J.; Gremeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variables | Spearman’s Correlation | p-Value |
|---|---|---|
| Total dietary fat intake (g) | 0.821 | 0.023 |
| Energy (kcal) | 0.929 | 0.003 |
| Dietary Cholesterol (mg) | 0.786 | 0.036 |
| Saturated fatty acids (g) | 0.857 | 0.014 |
| 10:0 Capric acid (g) | 0.786 | 0.036 |
| 16:0 Palmitic acid (g) | 0.893 | 0.007 |
| 18:0 Stearic acid (g) | 0.821 | 0.023 |
| Monounsat. fatty acids (g) | 0.821 | 0.023 |
| 18:1 Oleic Acid (g) | 0.821 | 0.023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholas, D.A.; Mbongue, J.C.; Garcia-Pérez, D.; Sorensen, D.; Bennit, H.F.; De Leon, M.; Langridge, W.H.R. Exploring the Interplay between Fatty Acids, Inflammation, and Type 2 Diabetes. Immuno 2024, 4, 91-107. https://doi.org/10.3390/immuno4010006
Nicholas DA, Mbongue JC, Garcia-Pérez D, Sorensen D, Bennit HF, De Leon M, Langridge WHR. Exploring the Interplay between Fatty Acids, Inflammation, and Type 2 Diabetes. Immuno. 2024; 4(1):91-107. https://doi.org/10.3390/immuno4010006
Chicago/Turabian StyleNicholas, Dequina A., Jacques C. Mbongue, Darysbel Garcia-Pérez, Dane Sorensen, Heather Ferguson Bennit, Marino De Leon, and William H. R. Langridge. 2024. "Exploring the Interplay between Fatty Acids, Inflammation, and Type 2 Diabetes" Immuno 4, no. 1: 91-107. https://doi.org/10.3390/immuno4010006