
The Importance of Neutrophils in Osteoarthritis: Current Concepts and Therapeutic Perspectives

 , , and
, , and 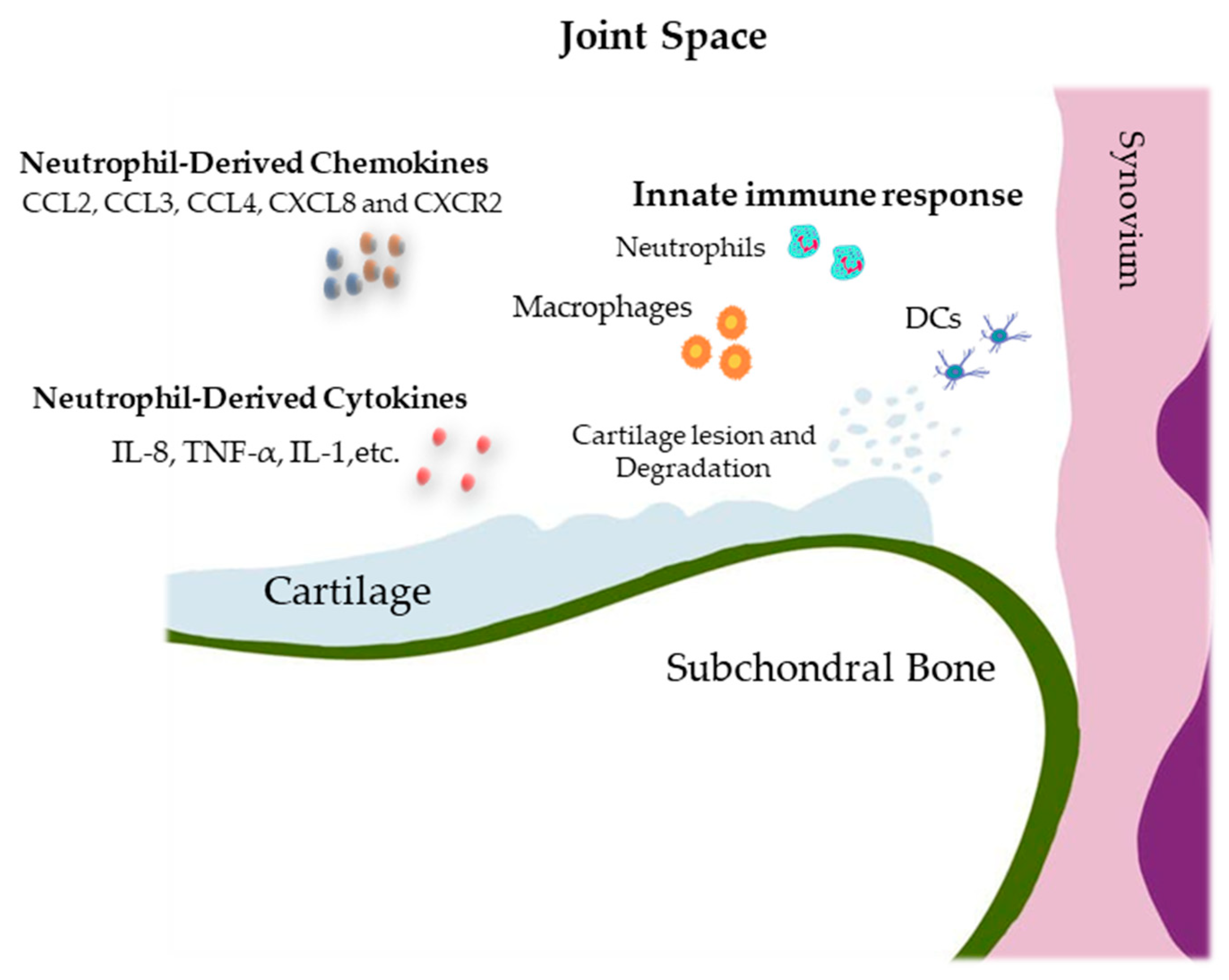{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Osteoarthritis
2.1. Osteoarthritis Epidemiology in Human
2.2. Pathogenesis
3. Involvement of Neutrophils in the Pathophysiology of Osteoarthritis
3.1. Immune Cells in Joint Injury and Repair
3.2. Trafficking of Circulating Blood Neutrophils to the Synovial Cavity
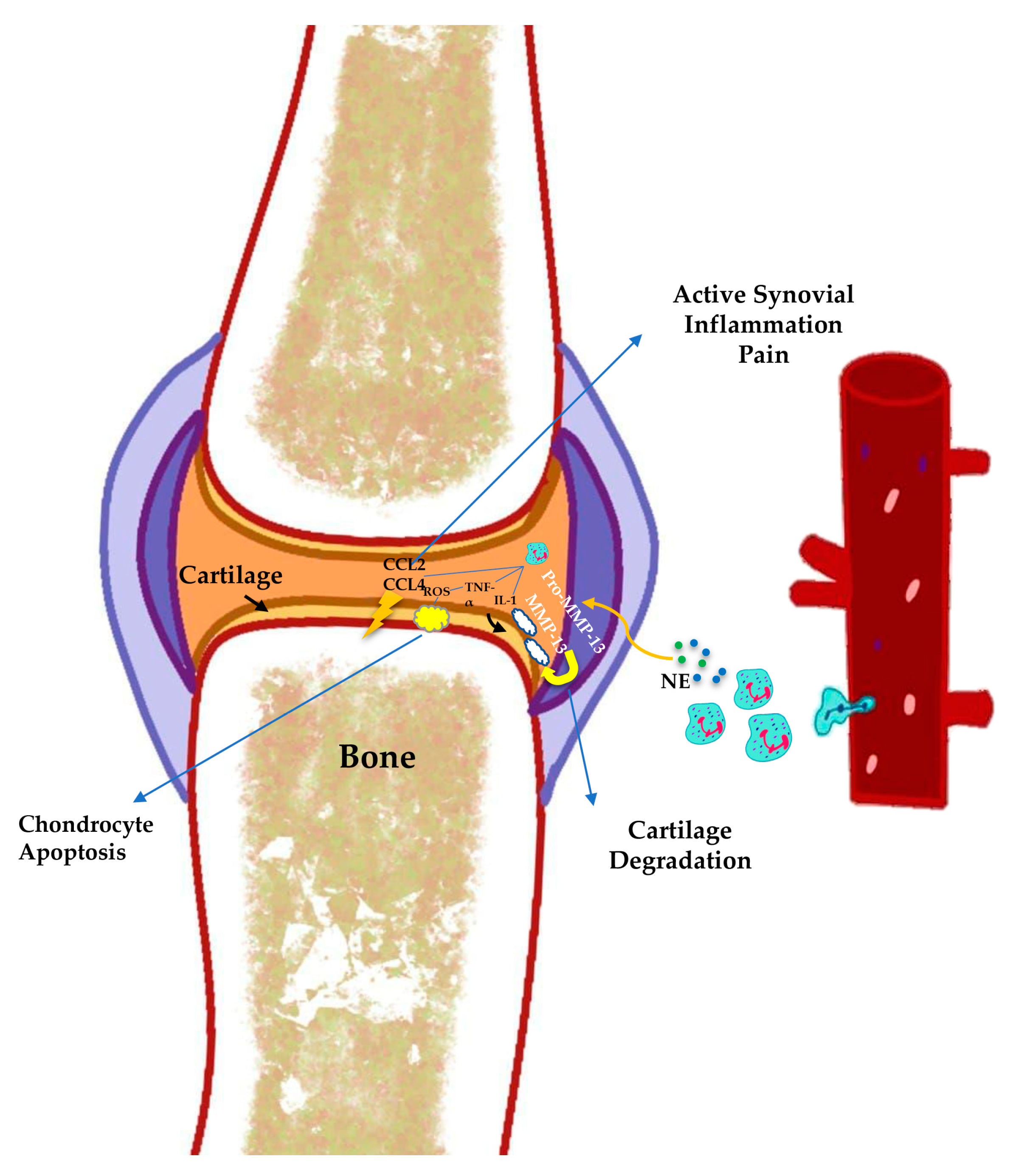
3.3. Neutrophil-Derived Cytokines, Chemokines, and Enzymes
3.4. Neutrophil Elastase
3.5. MicroRNAs Expression by Neutrophils
3.6. Neutrophils in Cartilage Degradation
3.7. Synovium, Cartilage, Subchondral Bone, and Innate Immunity
4. Neutrophil Biology, Recruitment, and Function in Inflammation
4.1. Neutrophil Life Cycle (Neutrophil Mobilization, Clearance, and Circulation)
4.2. Neutrophils in Acute and Chronic Inflammation
4.3. Neutrophil Crosstalk/Interaction with Other Circulation Cells (Platelets, Adaptative Immune Cells, Monocytes/Macrophages)
4.4. Neutrophil Heterogeneity in Chronic Inflammation
4.5. New Insights into NET in Inflammation
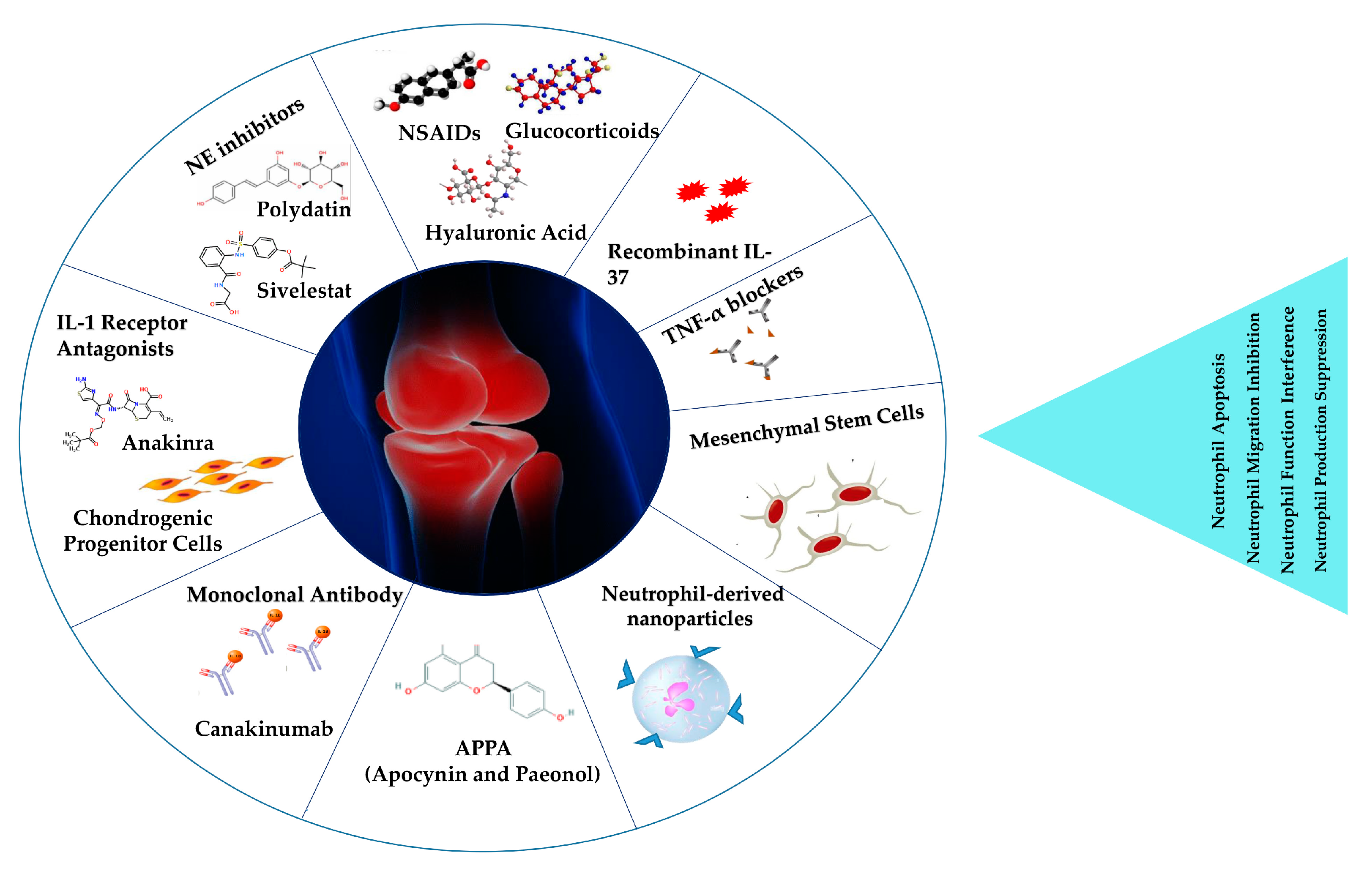
5. Neutrophils as a Target for Osteoarthritis Treatment
5.1. NSAIDs, Glucocorticoids, and Hyaluronic Acid
5.2. Anti-Cytokine Strategies
5.2.1. TNF-α Blockers
5.2.2. IL-1 and IL-6 Inhibitors
5.3. Recombinant IL-37
5.4. miRNA Modulation
5.5. NE and NET Inhibition
5.6. G-CSF Receptor Blockers
5.7. Biologic Therapy
5.8. Regenerative Medicine
5.9. Nanomedical Approaches in Osteoarthritis
Drug Delivery
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Senthelal, S.; Li, J.; Ardeshirzadeh, S.; Thomas, M.A. Arthritis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Drummer, D.; McAdam, J.; Seay, R.; Ferrando, A.; Bridges, S.L., Jr.; Singh, J.A.; Bamman, M. Osteoarthritis Progression: Mitigation and Rehabilitation Strategies. Front. Rehabil. Sci. 2021, 2, 724052. [Google Scholar] [CrossRef]
- Allen, K.; Thoma, L.; Golightly, Y. Epidemiology of osteoarthritis. Osteoarthr. Cartil. 2022, 30, 184–195. [Google Scholar] [CrossRef]
- Chaney, S.; Vergara, R.; Qiryaqoz, Z.; Suggs, K.; Akkouch, A. The Involvement of Neutrophils in the Pathophysiology and Treatment of Osteoarthritis. Biomedicines 2022, 10, 1604. [Google Scholar] [CrossRef]
- Molnar, V.; Matišić, V.; Kodvanj, I.; Bjelica, R.; Jeleč, Ž.; Hudetz, D.; Rod, E.; Čukelj, F.; Vrdoljak, T.; Vidović, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int. J. Mol. Sci. 2021, 22, 9208. [Google Scholar] [CrossRef]
- Sacitharan, P.K. Ageing and osteoarthritis. In Biochemistry and Cell Biology of Ageing: Part II Clinical Science; Springer: Berlin/Heidelberg, Germany, 2019; pp. 123–159. [Google Scholar]
- Radu, A.-F.; Bungau, S.G.; Tit, D.M.; Behl, T.; Uivaraseanu, B.; Marcu, M.F. Highlighting the Benefits of Rehabilitation Treatments in Hip Osteoarthritis. Medicina 2022, 58, 494. [Google Scholar] [CrossRef]
- Xu, M.; Ji, Y. Immunoregulation of synovial macrophages for the treatment of osteoarthritis. Open Life Sci. 2023, 18, 20220567. [Google Scholar] [CrossRef]
- Salman, L.A.; Ahmed, G.; Dakin, S.G.; Kendrick, B.; Price, A. Osteoarthritis: A narrative review of molecular approaches to disease management. Thromb. Haemost. 2023, 25, 27. [Google Scholar] [CrossRef]
- Grässel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Research 2020, 9, 325. [Google Scholar] [CrossRef]
- Arden, N.; Nevitt, M.C. Osteoarthritis: Epidemiology. Best Pract. Res. Clin. Rheumatol. 2006, 20, 3–25. [Google Scholar] [CrossRef]
- Curry, Z.A.M.; Beling, A.; Borg-Stein, J. Knee osteoarthritis in midlife women: Unique considerations and comprehensive management. Menopause 2022, 29, 748–755. [Google Scholar] [CrossRef]
- Tu, C.; He, J.; Wu, B.; Wang, W.; Li, Z. An extensive review regarding the adipokines in the pathogenesis and progression of osteoarthritis. Cytokine 2019, 113, 1–12. [Google Scholar] [CrossRef]
- Conway, B.; Rene, A. Obesity as a disease: No lightweight matter. Obes. Rev. 2004, 5, 145–151. [Google Scholar] [CrossRef]
- Ashkavand, Z.; Malekinejad, H.; Vishwanath, B.S. The pathophysiology of osteoarthritis. J. Pharm. Res. 2013, 7, 132–138. [Google Scholar] [CrossRef]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Thromb. Haemost. 2017, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jordan, J.M. Epidemiology of Osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, W.; Yong, H.; He, M.; Yang, Y.; Deng, Z.; Li, Y. Macrophages in osteoarthritis: Pathophysiology and therapeutics. Am. J. Transl. Res. 2020, 12, 261–268. [Google Scholar]
- Grässel, S.; Aszodi, A. Osteoarthritis and Cartilage Regeneration: Focus on Pathophysiology and Molecular Mechanisms. Int. J. Mol. Sci. 2019, 20, 6156. [Google Scholar] [CrossRef] [Green Version]
- Vincent, T.L. IL-1 in osteoarthritis: Time for a critical review of the literature. F1000Research 2019, 8, 934. [Google Scholar] [CrossRef]
- Mora, J.C.; Przkora, R.; Cruz-Almeida, Y. Knee osteoarthritis: Pathophysiology and current treatment modalities. J. Pain Res. 2018, 11, 2189–2196. [Google Scholar] [CrossRef] [Green Version]
- Liu-Bryan, R. Synovium and the Innate Inflammatory Network in Osteoarthritis Progression. Curr. Rheumatol. Rep. 2013, 15, 323. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yin, H.; Yan, Z.; Li, H.; Wu, J.; Wang, Y.; Wei, F.; Tian, G.; Ning, C.; Li, H.; et al. The immune microenvironment in cartilage injury and repair. Acta Biomater. 2022, 140, 23–42. [Google Scholar] [CrossRef]
- McDermott, J.E.; Pezzanite, L.; Goodrich, L.; Santangelo, K.; Chow, L.; Dow, S.; Wheat, W. Role of Innate Immunity in Initiation and Progression of Osteoarthritis, with Emphasis on Horses. Animals 2021, 11, 3247. [Google Scholar] [CrossRef]
- Arve-Butler, S.; Schmidt, T.; Mossberg, A.; Berthold, E.; Gullstrand, B.; Bengtsson, A.A.; Kahn, F.; Kahn, R. Synovial fluid neutrophils in oligoarticular juvenile idiopathic arthritis have an altered phenotype and impaired effector functions. Thromb. Haemost. 2021, 23, 109. [Google Scholar] [CrossRef]
- Fattori, V.; Amaral, F.A.; Verri, W.A. Neutrophils and arthritis: Role in disease and pharmacological perspectives. Pharmacol. Res. 2016, 112, 84–98. [Google Scholar] [CrossRef]
- Zdziennicka, J.; Szponder, T.; Wessely-Szponder, J. Application of Natural Neutrophil Products for Stimulation of Monocyte-Derived Macrophages Obtained before and after Osteochondral or Bone Injury. Microorganisms 2021, 9, 124. [Google Scholar] [CrossRef]
- Fujie, K.; Shinguh, Y.; Inamura, N.; Yasumitsu, R.; Okamoto, M.; Okuhara, M. Release of neutrophil elastase and its role in tissue injury in acute inflammation: Effect of the elastase inhibitor, FR134043. Eur. J. Pharmacol. 1999, 374, 117–125. [Google Scholar] [CrossRef]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2009, 339, 437–448. [Google Scholar] [CrossRef]
- Scanzello, C.R. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J. Orthop. Res. 2017, 35, 735–739. [Google Scholar] [CrossRef] [Green Version]
- López-Armada, M.J.; Carames, B.; Martín, M.A.; Cillero-Pastor, B.; Lires-Dean, M.; Fuentes-Boquete, I.; Arenas, J.; Blanco, F.J. Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthr. Cartil. 2006, 14, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Lehmann, C.; Bode, C.; Miosge, N.; Schubert, A. High Mobility Group Box 1 Protein in Osteoarthritic Knee Tissue and Chondrogenic Progenitor Cells: An Ex Vivo and In Vitro Study. Cartilage 2021, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119. [Google Scholar] [CrossRef] [Green Version]
- Blom, A.B.; van der Kraan, P.M.; van den Berg, W.B. Cytokine Targeting in Osteoarthritis. Curr. Drug Targets 2007, 8, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C. Alternative pathways of IL-1 activation, and its role in health and disease. Front. Immunol. 2020, 11, 3288. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Falconer, A.M.D.; Wright, H.L.; Lin, H.; Yamamoto, K.; Cheung, K.; Charlton, S.H.; Arques, M.D.C.; Janciauskiene, S.; Refaie, R.; et al. Matrix metalloproteinase-13 is fully activated by neutrophil elastase and inactivates its serpin inhibitor, alpha-1 antitrypsin: Implications for osteoarthritis. FEBS J. 2022, 289, 121–139. [Google Scholar] [CrossRef]
- Carrión, M.; Juarranz, Y.; Martínez, C.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology 2013, 52, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Deligne, C.; Casulli, S.; Pigenet, A.; Bougault, C.; Campillo-Gimenez, L.; Nourissat, G.; Berenbaum, F.; Elbim, C.; Houard, X. Differential expression of interleukin-17 and interleukin-22 in inflamed and non-inflamed synovium from osteoarthritis patients. Osteoarthr. Cartil. 2015, 23, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Grevers, L.C.; van Lent, P.L.E.M.; Koenders, M.I.; Walgreen, B.; Sloetjes, A.W.; Nimmerjahn, F.; Verbeek, J.S.; van den Berg, W.B. Different amplifying mechanisms of interleukin-17 and interferon-γ in Fcγ receptor–mediated cartilage destruction in murine immune complex–mediated arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 396–407. [Google Scholar] [CrossRef]
- Wang, G.; Jing, W.; Bi, Y.; Li, Y.; Ma, L.; Yang, H.; Zhang, Y. Neutrophil Elastase Induces Chondrocyte Apoptosis and Facilitates the Occurrence of Osteoarthritis via Caspase Signaling Pathway. Front. Pharmacol. 2021, 12, 666162. [Google Scholar] [CrossRef]
- Muley, M.M.; Reid, A.R.; Botz, B.; Bölcskei, K.; Helyes, Z.; McDougall, J.J. Neutrophil elastase induces inflammation and pain in mouse knee joints via activation of proteinase-activated receptor-2. Br. J. Pharmacol. 2016, 173, 766–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneva, M.K. Neutrophil elastase and its inhibitors—Overlooked players in osteoarthritis. FEBS J. 2022, 289, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Prajzlerová, K.; Kryštůfková, O.; Hánová, P.; Horváthová, V.; Gregová, M.; Pavelka, K.; Vencovský, J.; Šenolt, L.; Filková, M. High miR-451 expression in peripheral blood mononuclear cells from subjects at risk of developing rheumatoid arthritis. Sci. Rep. 2021, 11, 4719. [Google Scholar] [CrossRef] [PubMed]
- Águila, S.; Reyes-García, A.M.d.L.; Fernández-Pérez, M.P.; Reguilón-Gallego, L.; Zapata-Martínez, L.; Ruiz-Lorente, I.; Vicente, V.; González-Conejero, R.; Martínez, C. Micrornas as new regulators of neutrophil extracellular trap formation. Int. J. Mol. Sci. 2021, 22, 2116. [Google Scholar] [CrossRef]
- Gurol, T.; Zhou, W.; Deng, Q. Micro RNA s in neutrophils: Potential next generation therapeutics for inflammatory ailments. Immunol. Rev. 2016, 273, 29–47. [Google Scholar] [CrossRef]
- de Almeida, R.C.; Ramos, Y.F.M.; Mahfouz, A.; den Hollander, W.; Lakenberg, N.; Houtman, E.; van Hoolwerff, M.; Suchiman, H.E.D.; Ruiz, A.R.; Slagboom, P.E.; et al. RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis. 2019, 78, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Okuhara, A.; Nakasa, T.; Shibuya, H.; Niimoto, T.; Adachi, N.; Deie, M.; Ochi, M. Changes in microRNA expression in peripheral mononuclear cells according to the progression of osteoarthritis. Mod. Rheumatol. 2012, 22, 446–457. [Google Scholar] [CrossRef]
- McClurg, O.; Tinson, R.; Troeberg, L. Targeting Cartilage Degradation in Osteoarthritis. Pharmaceuticals 2021, 14, 126. [Google Scholar] [CrossRef]
- Majtnerová, P.; Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 2018, 45, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, Q.; Yang, M.; Yang, L.; Wang, W.; Ding, H.; Zhang, D.; Xu, J.; Tang, X.; Ding, H.; et al. Histomorphology and innate immunity during the progression of osteoarthritis: Does synovitis affect cartilage degradation? J. Cell. Physiol. 2018, 233, 1342–1358. [Google Scholar] [CrossRef]
- Wilson, M.E.; Mccandless, E.E.; Olszewski, M.A.; Robinson, N.E. VA Ann Arbor Healthcare System. Res. Serv. 2020, 256, 105436. [Google Scholar] [CrossRef]
- Hsueh, M.; Zhang, X.; Wellman, S.S.; Bolognesi, M.; Kraus, V.B. Synergistic Roles of Macrophages and Neutrophils in Osteoarthritis Progression. Arthritis Rheumatol. 2021, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Haraden, C.A.; Huebner, J.L.; Hsueh, M.-F.; Li, Y.-J.; Kraus, V.B. Synovial fluid biomarkers associated with osteoarthritis severity reflect macrophage and neutrophil related inflammation. Thromb. Haemost. 2019, 21, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, V.; McDaniel, G.; Huebner, J.; Stabler, T.; Pieper, C.; Shipes, S.; Petry, N.; Low, P.; Shen, J.; McNearney, T.; et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthr. Cartil. 2016, 24, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hunter, D.; Jin, X.; Ding, C. The importance of synovial inflammation in osteoarthritis: Current evidence from imaging assessments and clinical trials. Osteoarthr. Cartil. 2018, 26, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, A.; Hilkens, C.M. Synovial Macrophages in Osteoarthritis: The Key to Understanding Pathogenesis? Front. Immunol. 2021, 12, 678757. [Google Scholar] [CrossRef]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlwraith, C.W. Traumatic arthritis and posttraumatic osteoarthritis in the horse. In Joint Disease in the Horse; Elsevier: Amsterdam, The Netherlands, 2016; pp. 33–48. [Google Scholar]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Goldring, S.R. Alterations in periarticular bone and cross talk between subchondral bone and articular cartilage in osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Benigni, G.; Dimitrova, P.; Antonangeli, F.; Sanseviero, E.; Milanova, V.; Blom, A.; van Lent, P.; Morrone, S.; Santoni, A.; Bernardini, G. CXCR3/CXCL10 axis regulates neutrophil–NK cell cross-talk determining the severity of experimental osteoarthritis. J. Immunol. 2017, 198, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.D.; Kent, C.L.; Huebner, J.L.; Kraus, V.B.; McNulty, A.L.; Guilak, F.; Olson, S.A. CXCL10 is upregulated in synovium and cartilage following articular fracture. J. Orthop. Res. 2018, 36, 1220–1227. [Google Scholar] [CrossRef]
- Jung, Y.-K.; Han, M.-S.; Park, H.-R.; Lee, E.-J.; Jang, J.-A.; Kim, G.-W.; Lee, S.-Y.; Moon, D.; Han, S. Calcium-phosphate complex increased during subchondral bone remodeling affects earlystage osteoarthritis. Sci. Rep. 2018, 8, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple Roles for Chemokines in Neutrophil Biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef]
- Lerman, Y.V.; Kim, M. Neutrophil migration under normal and sepsis conditions. Cardiovasc. Haematol. Disord. Drug Targets 2015, 15, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef]
- Mócsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Burn, G.L.; Foti, A.; Marsman, G.; Patel, D.F.; Zychlinsky, A. The neutrophil. Immunity 2021, 54, 1377–1391. [Google Scholar] [CrossRef]
- Christoffersson, G.; Phillipson, M. The neutrophil: One cell on many missions or many cells with different agendas? Cell Tissue Res. 2018, 371, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furze, R.C.; Rankin, S.M. The role of the bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J. 2008, 22, 3111–3119. [Google Scholar] [CrossRef]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef] [Green Version]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: Insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef]
- Wright, H.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Gupta, K.; Shukla, M.; Cowland, J.B.; Malemud, C.J.; Haqqi, T.M. Neutrophil gelatinase–associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007, 56, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.M.; Betz, T.V.; Lamont, D.J.; Kim, M.B.; Shaw, S.K.; Froio, R.M.; Baleux, F.; Arenzana-Seisdedos, F.; Alon, R.; Luscinskas, F.W. Elastase Release by Transmigrating Neutrophils Deactivates Endothelial-bound SDF-1α and Attenuates Subsequent T Lymphocyte Transendothelial Migration. J. Exp. Med. 2004, 200, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1986, 29, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. Phagocytosis, Degranulation and Extracellular Traps Release by Neutrophils—The Current Knowledge, Pharmacological Modulation and Future Prospects. Front. Pharmacol. 2021, 12, 666732. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb. Haemost. 2018, 118, 006–027. [Google Scholar] [CrossRef] [Green Version]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Headland, S.E.; Jones, H.R.; Norling, L.V.; Kim, A.; Souza, P.R.; Corsiero, E.; Gil, C.D.; Nerviani, A.; Dell’accio, F.; Pitzalis, C.; et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 2015, 7, 315ra190. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G. Targeting Neutrophils for Promoting the Resolution of Inflammation. Front. Immunol. 2022, 13, 1008. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.-P.; Martel-Pelletier, J.; Rannou, F.; Cooper, C. Efficacy and safety of oral NSAIDs and analgesics in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin. Arthritis Rheum. 2015, 45, S22–S27. [Google Scholar] [CrossRef] [Green Version]
- Paglia, M.D.G.; Silva, M.T.; Lopes, L.C.; Barberato-Filho, S.; Mazzei, L.G.; Abe, F.C.; Bergamaschi, C.D.C. Use of corticoids and non-steroidal anti-inflammatories in the treatment of rheumatoid arthritis: Systematic review and network meta-analysis. PLoS ONE 2021, 16, e0248866. [Google Scholar] [CrossRef]
- Bertolotto, M.; Contini, P.; Ottonello, L.; Pende, A.; Dallegri, F.; Montecucco, F. Neutrophil migration towards C5a and CXCL8 is prevented by non-steroidal anti-inflammatory drugs via inhibition of different pathways. Br. J. Pharmacol. 2014, 171, 3376–3393. [Google Scholar] [CrossRef] [Green Version]
- Marsolais, D.; Côté, C.H.; Frenette, J. Nonsteroidal Anti-Inflammatory Drug Reduces Neutrophil and Macrophage Accumulation but Does Not Improve Tendon Regeneration. Lab. Investig. 2003, 83, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Topete, D.; Cidlowski, J.A. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Xu, K.; Liang, Y.; Du, P.; Wan, S.; Jiang, C. Effect of hyaluronic acid on cytokines and immune cells change in patients of knee osteoarthritis. BMC Musculoskelet. Disord. 2022, 23, 812. [Google Scholar] [CrossRef]
- Nicholls, M.; Fierlinger, A.; Niazi, F.; Bhandari, M. The Disease-Modifying Effects of Hyaluronan in the Osteoarthritic Disease State. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2017, 10, 1179544117723611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Wang, C.-T.; Chou, W.-C.; Kao, C.-L.; Tsa, K.-L. Hyaluronic acid injection reduces inflammatory and apoptotic markers through modulation of AKT by repressing the oxidative status of neutrophils from osteoarthritic synovial fluid. Int. J. Biol. Macromol. 2020, 165, 2765–2772. [Google Scholar] [CrossRef]
- Dinarello, C.A. Anti-inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Deng, Z.; Chen, K.; Jian, S.; Zhou, F.; Yang, Y.; Fu, Z.; Xie, H.; Xiong, J.; Zhu, W. Cartilage tissue engineering: From proinflammatory and anti-inflammatory cytokines to osteoarthritis treatments (Review). Mol. Med. Rep. 2022, 25, 99. [Google Scholar] [CrossRef] [PubMed]
- Chisari, E.; Yaghmour, K.; Khan, W. The effects of TNF-alpha inhibition on cartilage: A systematic review of preclinical studies. Osteoarthr. Cartil. 2020, 28, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-R.; Yoo, J.J.; Kim, H.A. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shi, N.; Diao, Z.; Chen, Y.; Zhang, Y. Therapeutic potential of TNFα inhibitors in chronic inflammatory disorders: Past and future. Genes Dis. 2020, 8, 38–47. [Google Scholar] [CrossRef]
- Hastings, R.; Ding, T.; Butt, S.; Gadsby, K.; Zhang, W.; Moots, R.J.; Deighton, C. Neutropenia in patients receiving anti-tumor necrosis factor therapy. Arthritis Care Res. 2010, 62, 764–769. [Google Scholar] [CrossRef]
- Zhang, C.; Shu, W.; Zhou, G.; Lin, J.; Chu, F.; Wu, H.; Liu, Z. Anti-TNF-α Therapy Suppresses Proinflammatory Activities of Mucosal Neutrophils in Inflammatory Bowel Disease. Mediat. Inflamm. 2018, 2018, 3021863. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef] [Green Version]
- Potera, R.M.; Jensen, M.J.; Hilkin, B.M.; South, G.K.; Hook, J.S.; Gross, E.A.; Moreland, J.G. Neutrophil azurophilic granule exocytosis is primed by TNF-α and partially regulated by NADPH oxidase. J. Endotoxin Res. 2016, 22, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capsoni, F.; Sarzi-Puttini, P.; Atzeni, F.; Minonzio, F.; Bonara, P.; Doria, A.; Carrabba, M. Effect of adalimumab on neutrophil function in patients with rheumatoid arthritis. Thromb. Haemost. 2005, 7, R250–R255. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.C.; Peters, A.M.; Paleolog, E.; Chapman, P.T.; Elliott, M.J.; McCloskey, R.; Feldmann, M.; Maini, R.N. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor α blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 38–47. [Google Scholar] [CrossRef]
- Chevalier, X.; Eymard, F. Anti-IL-1 for the treatment of OA: Dead or alive? Nat. Rev. Rheumatol. 2019, 15, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Ronacher, L.; Liu-Bryan, R.; Takai, S.; Karin, M.; Corr, M. Caspase 1–independent activation of interleukin-1β in neutrophil-predominant inflammation. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 3642–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology 2004, 43, iii2–iii9. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009, 61, 344–352. [Google Scholar] [CrossRef]
- Prince, L.R.; Allen, L.; Jones, E.C.; Hellewell, P.G.; Dower, S.K.; Whyte, M.K.; Sabroe, I. The Role of Interleukin-1β in Direct and Toll-Like Receptor 4-Mediated Neutrophil Activation and Survival. Am. J. Pathol. 2004, 165, 1819–1826. [Google Scholar] [CrossRef]
- Özcan, A.; Boyman, O. Mechanisms regulating neutrophil responses in immunity, allergy, and autoimmunity. Allergy 2022, 77, 3567–3583. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Akhtar, S.; Porter, R.; Onnerfjord, P.; Bajpayee, A.G. Interleukin-1 receptor antagonist (IL-1Ra) is more effective in suppressing cytokine-induced catabolism in cartilage-synovium co-culture than in cartilage monoculture. Thromb. Haemost. 2019, 21, 238. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Koenders, M.; Kalabokis, V.; Kim, J.; Tan, A.C.; Garlanda, C.; Mantovani, A.; Dagna, L.; Joosten, L.A.B.; Dinarello, C.A. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatology 2016, 55, 2220–2229. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.; Carmona-Rivera, C.; Ombrello, A.K.; Hoffmann, P.; Seto, N.L.; Jones, A.; Stone, D.L.; Naz, F.; Carlucci, P.; Dell’orso, S.; et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann. Rheum. Dis. 2018, 77, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yuan, S.; Zeng, Y.; Wang, C.; Yu, N.; Ding, C. New Trends in Pharmacological Treatments for Osteoarthritis. Front. Pharmacol. 2021, 12, 645842. [Google Scholar] [CrossRef]
- Cheleschi, S.; Cantarini, L.; Pascarelli, N.A.; Collodel, G.; Lucherini, O.M.; Galeazzi, M.; Fioravanti, A. Possible chondroprotective effect of canakinumab: An in vitro study on human osteoarthritic chondrocytes. Cytokine 2015, 71, 165–172. [Google Scholar] [CrossRef]
- Ghouri, A.; Conaghan, P.G. Prospects for Therapies in Osteoarthritis. Calcif. Tissue Int. 2021, 109, 339–350. [Google Scholar] [CrossRef]
- Ohyama, A.; Osada, A.; Kawaguchi, H.; Kurata, I.; Nishiyama, T.; Iwai, T.; Ishigami, A.; Kondo, Y.; Tsuboi, H.; Sumida, T.; et al. Specific Increase in Joint Neutrophil Extracellular Traps and Its Relation to Interleukin 6 in Autoimmune Arthritis. Int. J. Mol. Sci. 2021, 22, 7633. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-M.; Meng, H.-Y.; Yuan, X.-L.; Wang, Y.; Guo, Q.-Y.; Peng, J.; Wang, A.-Y.; Lu, S.-B. MicroRNAs’ Involvement in Osteoarthritis and the Prospects for Treatments. Evid.-Based Complement. Altern. Med. 2015, 2015, 236179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malemud, C.J. MicroRNAs and Osteoarthritis. Cells 2018, 7, 92. [Google Scholar] [CrossRef]
- Lu, X.; Li, Y.; Chen, H.; Pan, Y.; Lin, R.; Chen, S. miR-335-5P contributes to human osteoarthritis by targeting HBP1. Exp. Ther. Med. 2021, 21, 109. [Google Scholar] [CrossRef]
- Kmiołek, T.; Paradowska-Gorycka, A. miRNAs as Biomarkers and Possible Therapeutic Strategies in Rheumatoid Arthritis. Cells 2022, 11, 452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, C.; Zhang, C.; Luo, C.; Zhong, B.; Yu, X. MiRNA-132 regulates the development of osteoarthritis in correlation with the modulation of PTEN/PI3K/AKT signaling. BMC Geriatr. 2021, 21, 175. [Google Scholar] [CrossRef]
- Castro-Villegas, C.; Pérez-Sánchez, C.; Escudero, A.; Filipescu, I.; Verdu, M.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gomez, Y.; Font, P.; Rodriguez-Ariza, A.; et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Thromb. Haemost. 2015, 17, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Rosa, I.A.; Perez-Sanchez, C.; Ruiz-Limon, P.; Patiño-Trives, A.; Torres-Granados, C.; Jimenez-Gomez, Y.; Abalos-Aguilera, M.D.C.; Cecchi, I.; Ortega, R.; Caracuel, M.A.; et al. Impaired microRNA processing in neutrophils from rheumatoid arthritis patients confers their pathogenic profile. Modulation by biological therapies. Haematologica 2020, 105, 2250. [Google Scholar] [CrossRef] [Green Version]
- Muley, M.M.; Krustev, E.; Reid, A.R.; McDougall, J.J. Prophylactic inhibition of neutrophil elastase prevents the development of chronic neuropathic pain in osteoarthritic mice. J. Neuroinflamm. 2017, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, L.D.C.; Micheli, L.; Cinci, L.; Maresca, M.; Vergelli, C.; Pacini, A.; Quinn, M.T.; Giovannoni, M.P.; Ghelardini, C. Effects of the neutrophil elastase inhibitor EL-17 in rat adjuvant-induced arthritis. Rheumatology 2016, 55, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhao, L.; Yu, Z.; Yu, C.; Bi, J.; Sun, B.; Cong, H. Sivelestat sodium hydrate improves post-traumatic knee osteoarthritis through nuclear factor-κB in a rat model. Exp. Ther. Med. 2017, 14, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocetti, L.; Giovannoni, M.P.; Cantini, N.; Guerrini, G.; Vergelli, C.; Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. Novel Sulfonamide Analogs of Sivelestat as Potent Human Neutrophil Elastase Inhibitors. Front. Chem. 2020, 8, 795. [Google Scholar] [CrossRef]
- Tadie, J.-M.; Bae, H.-B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.-F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Ming, B.; Dong, L. The Role of HMGB1 in Rheumatic Diseases. Front. Immunol. 2022, 13, 815257. [Google Scholar] [CrossRef]
- Li, R.-L.; Duan, H.-X.; Liang, Q.; Huang, Y.-L.; Wang, L.-Y.; Zhang, Q.; Wu, C.-J.; Liu, S.-Q.; Peng, W. Targeting matrix metalloproteases: A promising strategy for herbal medicines to treat rheumatoid arthritis. Front. Immunol. 2022, 13, 1046810. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Clarke, J. NETs directly injure cartilage in RA. Nat. Rev. Rheumatol. 2020, 16, 410. [Google Scholar] [CrossRef]
- Yang, F.; Luo, X.; Luo, G.; Zhai, Z.; Zhuang, J.; He, J.; Han, J.; Zhang, Y.; Zhuang, L.; Sun, E.; et al. Inhibition of NET formation by polydatin protects against collagen-induced arthritis. Int. Immunopharmacol. 2019, 77, 105919. [Google Scholar] [CrossRef]
- Semerad, C.L.; Liu, F.; Gregory, A.D.; Stumpf, K.; Link, D.C. G-CSF Is an Essential Regulator of Neutrophil Trafficking from the Bone Marrow to the Blood. Immunity 2002, 17, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; Mackenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic Targeting of the G-CSF Receptor Reduces Neutrophil Trafficking and Joint Inflammation in Antibody-Mediated Inflammatory Arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.J.; Xie, L.; Ang, C.; Fahimi, F.; Willingham, S.B.; Kueh, A.J.; Herold, M.J.; Mackay, C.R.; Robert, R. Therapeutic blockade of CXCR2 rapidly clears inflammation in arthritis and atopic dermatitis models: Demonstration with surrogate and humanized antibodies. MAbs 2020, 12, 1856460. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Tanaka, Y. Progress in understanding the safety and efficacy of Janus kinase inhibitors for treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2016, 12, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Larkins, N.; King, C.M. Effectiveness of apocynin-paeonol (APPA) for the management of osteoarthritis in dogs: Comparisons with placebo and meloxicam in client-owned dogs. Matters 2017, 3, e201608000001. [Google Scholar] [CrossRef] [Green Version]
- Cross, A.L.; Hawkes, J.; Wright, H.L.; Moots, R.J.; Edwards, S.W. APPA (apocynin and paeonol) modulates pathological aspects of human neutrophil function, without supressing antimicrobial ability, and inhibits TNFα expression and signalling. Inflammopharmacology 2020, 28, 1223–1235. [Google Scholar] [CrossRef]
- Han, S. Osteoarthritis year in review 2022: Biology. Osteoarthr. Cartil. 2022, 30, 1575–1582. [Google Scholar] [CrossRef]
- Freitag, J.; Bates, D.; Boyd, R.; Shah, K.; Barnard, A.; Huguenin, L.; Tenen, A. Mesenchymal stem cell therapy in the treatment of osteoarthritis: Reparative pathways, safety and efficacy—A review. BMC Musculoskelet. Disord. 2016, 17, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maumus, M.; Guérit, D.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal stem cell-based therapies in regenerative medicine: Applications in rheumatology. Stem Cell Res. Ther. 2011, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wu, W.; Qu, X. Mesenchymal stem cells in osteoarthritis therapy: A review. Am. J. Transl. Res. 2021, 13, 448–461. [Google Scholar]
- Le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 168, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Castillo, M.; Liu, K.; Bonilla, L.; Rameshwar, P. The immune properties of mesenchymal stem cells. Int. J. Biomed. Sci. IJBS 2007, 3, 76. [Google Scholar]
- Jiang, W.; Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2019, 53, e12712. [Google Scholar] [CrossRef] [Green Version]
- Joel, M.D.M.; Yuan, J.; Wang, J.; Yan, Y.; Qian, H.; Zhang, X.; Xu, W.; Mao, F. MSC: Immunoregulatory effects, roles on neutrophils and evolving clinical potentials. Am. J. Transl. Res. 2019, 11, 3890–3904. [Google Scholar] [PubMed]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Molnar, V.; Pavelić, E.; Vrdoljak, K.; Čemerin, M.; Klarić, E.; Matišić, V.; Bjelica, R.; Brlek, P.; Kovačić, I.; Tremolada, C.; et al. Mesenchymal Stem Cell Mechanisms of Action and Clinical Effects in Osteoarthritis: A Narrative Review. Genes 2022, 13, 949. [Google Scholar] [CrossRef]
- Planat-Benard, V.; Varin, A.; Casteilla, L. MSCs and Inflammatory Cells Crosstalk in Regenerative Medicine: Concerted Actions for Optimized Resolution Driven by Energy Metabolism. Front. Immunol. 2021, 12, 626755. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Djonov, V.; Volarevic, V. The Cross-Talk between Mesenchymal Stem Cells and Immune Cells in Tissue Repair and Regeneration. Int. J. Mol. Sci. 2021, 22, 2472. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Z.; Bai, L. Cell interplay in osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 720477. [Google Scholar] [CrossRef]
- Dai, H.; Chen, R.; Gui, C.; Tao, T.; Ge, Y.; Zhao, X.; Qin, R.; Yao, W.; Gu, S.; Jiang, Y.; et al. Eliminating senescent chondrogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 2020, 11, 199. [Google Scholar] [CrossRef]
- Ding, L.; Zhou, C.; Zheng, H.; Wang, Q.; Song, H.; Buckwalter, J.A.; Martin, J.A. Migrating Progenitor Cells Derived From Injured Cartilage Surface Respond to Damage-Associated Molecular Patterns. Cartilage 2021, 13, 755S–765S. [Google Scholar] [CrossRef]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018, 13, 1182–1190. [Google Scholar] [CrossRef]
- Liu, L.; Pan, D.; Chen, S.; Martikainen, M.-V.; Kårlund, A.; Ke, J.; Pulkkinen, H.; Ruhanen, H.; Roponen, M.; Käkelä, R.; et al. Systematic design of cell membrane coating to improve tumor targeting of nanoparticles. Nat. Commun. 2022, 13, 6181. [Google Scholar] [CrossRef]
- Narain, A.; Asawa, S.; Chhabria, V.; Patil-Sen, Y. Cell membrane coated nanoparticles: Next-generation therapeutics. Nanomedicine 2017, 12, 2677–2692. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, B.; Guo, J.; Zheng, L.; Guo, Q.; Peng, J.; Xu, J.; Qin, L. Nanoparticle–Cartilage Interaction: Pathology-Based Intra-articular Drug Delivery for Osteoarthritis Therapy. Nano-Micro Lett. 2021, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Collison, J. Nanoparticles in neutrophil clothing. Nat. Rev. Rheumatol. 2018, 14, 622. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Z.; Pang, Y.; Zhou, H. The interaction between nanoparticles and immune system: Application in the treatment of inflammatory diseases. J. Nanobiotechnol. 2022, 20, 127. [Google Scholar] [CrossRef]
- Zhang, Q.; Gong, H.; Gao, W.; Zhang, L. Recent Progress in Capturing and Neutralizing Inflammatory Cytokines. CCS Chem. 2020, 2, 376–389. [Google Scholar] [CrossRef]
- Yu, Q.; Huang, Y.; Chen, X.; Chen, Y.; Zhu, X.; Liu, Y.; Liu, J. A neutrophil cell membrane-biomimetic nanoplatform based on l-arginine nanoparticles for early osteoarthritis diagnosis and nitric oxide therapy. Nanoscale 2022, 14, 11619–11634. [Google Scholar] [CrossRef]
- Wang, H.; Zang, J.; Zhao, Z.; Zhang, Q.; Chen, S. The Advances of Neutrophil-Derived Effective Drug Delivery Systems: A Key Review of Managing Tumors and Inflammation. Int. J. Nanomed. 2021, 16, 7663–7681. [Google Scholar] [CrossRef]
- Su, Y.; Gao, J.; Kaur, P.; Wang, Z. Neutrophils and Macrophages as Targets for Development of Nanotherapeutics in Inflammatory Diseases. Pharmaceutics 2020, 12, 1222. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef]
- Marino, A.A.; Waddell, D.D.; Kolomytkin, O.V.; Meek, W.D.; Wolf, R.; Sadasivan, K.K.; Albright, J.A. Increased Intercellular Communication through Gap Junctions May Contribute to Progression of Osteoarthritis. Clin. Orthop. Relat. Res. 2004, 422, 224–232. [Google Scholar] [CrossRef]
- Carpintero-Fernandez, P.; Gago-Fuentes, R.; Wang, H.Z.; Fonseca, E.; Caeiro, J.R.; Valiunas, V.; Brink, P.R.; Mayan, M.D. Intercellular communication via gap junction channels between chondrocytes and bone cells. Biochim. Biophys. Acta BBA—Biomembr. 2018, 1860, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, G.B.; Bunn, K.E.; Pua, H.H.; Rafat, M. Extracellular vesicles: Mediators of intercellular communication in tissue injury and disease. Cell Commun. Signal. 2021, 19, 104. [Google Scholar] [CrossRef] [PubMed]
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exosomes: Insights From Exercise Science. Front. Physiol. 2021, 11, 604274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, X.; Zhao, J.; Yang, Y.; Cai, X.; Xu, J.; Cao, P. Exosomes: A Novel Strategy for Treatment and Prevention of Diseases. Front. Pharmacol. 2017, 8, 300. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bréchard, S. Neutrophil Extracellular Vesicles: A Delicate Balance between Pro-Inflammatory Responses and Anti-Inflammatory Therapies. Cells 2022, 11, 3318. [Google Scholar] [CrossRef]
- Rondelli, V.; Helmy, S.; Passignani, G.; Parisse, P.; Di Silvestre, D. Integrated Strategies for a Holistic View of Extracellular Vesicles. ACS Omega 2022, 7, 19058–19069. [Google Scholar] [CrossRef]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord.-Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Qin, Z.; Sun, H.; Chen, X.; Dong, J.; Shen, S.; Zheng, L.; Gu, N.; Jiang, Q. Nanoenzyme engineered neutrophil-derived exosomes attenuate joint injury in advanced rheumatoid arthritis via regulating inflammatory environment. Bioact. Mater. 2022, 18, 1–14. [Google Scholar] [CrossRef]
- Hong, C.-W. Extracellular Vesicles of Neutrophils. Immune Netw. 2018, 18, e43. [Google Scholar] [CrossRef]
- Zhan, D.; Cross, A.; Wright, H.L.; Moots, R.J.; Edwards, S.W.; Honsawek, S. Internalization of Neutrophil-Derived Microvesicles Modulates TNFα-Stimulated Proinflammatory Cytokine Production in Human Fibroblast-Like Synoviocytes. Int. J. Mol. Sci. 2021, 22, 7409. [Google Scholar] [CrossRef]
- Yang, N.; Li, M.; Wu, L.; Song, Y.; Yu, S.; Wan, Y.; Cheng, W.; Yang, B.; Mou, X.; Yu, H.; et al. Peptide-anchored neutrophil membrane-coated biomimetic nanodrug for targeted treatment of rheumatoid arthritis. J. Nanobiotechnol. 2023, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Miller, R.J.; Malfait, A.-M. Osteoarthritis joint pain: The cytokine connection. Cytokine 2014, 70, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehrani, Y.; Rahimi Junqani, R.; Morovati, S.; Mehrani, H.; Karimi, N.; Ghasemi, S. The Importance of Neutrophils in Osteoarthritis: Current Concepts and Therapeutic Perspectives. Immuno 2023, 3, 250-272. https://doi.org/10.3390/immuno3030017
Mehrani Y, Rahimi Junqani R, Morovati S, Mehrani H, Karimi N, Ghasemi S. The Importance of Neutrophils in Osteoarthritis: Current Concepts and Therapeutic Perspectives. Immuno. 2023; 3(3):250-272. https://doi.org/10.3390/immuno3030017
Chicago/Turabian StyleMehrani, Yeganeh, Rasool Rahimi Junqani, Solmaz Morovati, Hossein Mehrani, Negar Karimi, and Samaneh Ghasemi. 2023. "The Importance of Neutrophils in Osteoarthritis: Current Concepts and Therapeutic Perspectives" Immuno 3, no. 3: 250-272. https://doi.org/10.3390/immuno3030017