
Polymorphonuclear Neutrophils in Rheumatoid Arthritis and Systemic Lupus Erythematosus: More Complicated Than Anticipated


Abstract
:1. Introduction
2. Interaction of PMNs with Other Immune Cells and Non-Classical Functions of PMNs
3. Neutrophil Extracellular Trap Formation, Function and Clearance
4. Involvement of PMNs in RA and SLE
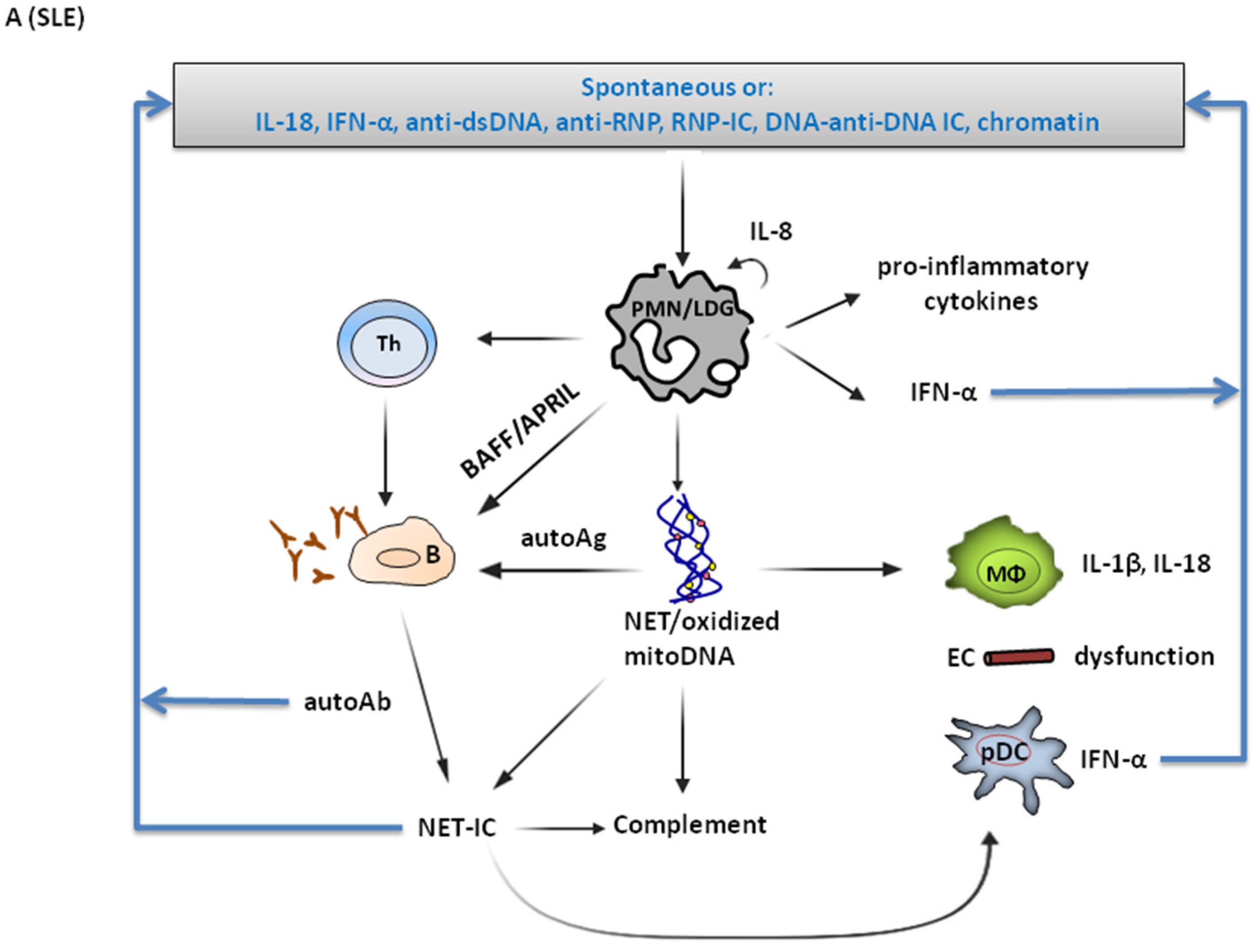
4.1. Involvement in SLE Pathogenesis
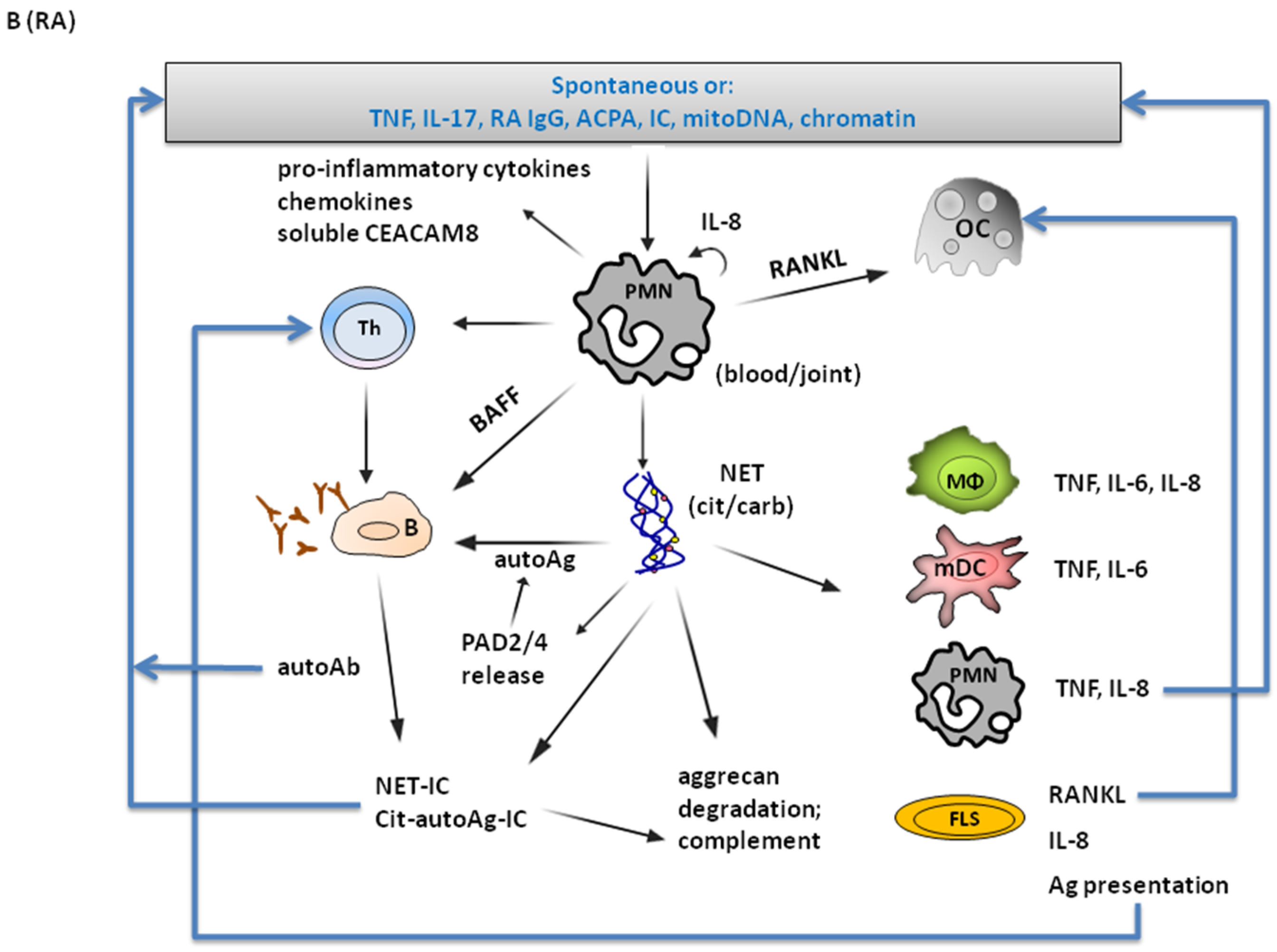
4.2. Involvement in RA Pathogenesis

{kind=link}
{kind=link}
| SLE | RA | |
|---|---|---|
| PMNs are activated | [79] | [80] |
| PMNs detected in affected tissues | [82] | [83,84] |
| Pro-inflammatory LDGs 1 | [10] | [129] |
| Altered PMN phenotype | [87,88] | [80,120,121,123,126] |
| PMNs express/produce key disease cytokines | IFN-α [28] | TNF [123] |
| SLE | RA | |
|---|---|---|
| Higher NET 1 formation | [82] | [42,130,131] |
| Impaired NET clearance | [75,102] | [144] |
| Anti-NET antibodies | [75,85,102] | [85] |
| NETs detected in affected tissues | Skin, kidney [82] | Rheumatoid nodules [130] Synovial fluid [132] |
| Disease-specific antibodies bind NETs | Anti-dsDNA [114] | ACPA-rich IgGs [42,130] Purified ACPAs [133] |
| Disease-specific antibodies induce NETs | Anti-dsDNA [98] | ACPA-rich IgGs [130] Purified ACPAs [133] |
| NETs directly activate immune cells or key cells involved in disease pathogenicity | Immune cells [103,104,106,107,108] Endothelial cells [105] | Immune cells [42,131,137] FLSs [130,137] |
| NETs in immune complexes activate immune cells | [106] | [139] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pillay, J.; Den Braber, I.; Vrisekoop, N.; Kwast, L.M.; De Boer, R.J.; Borghans, J.A.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef]
- Dixon, G.; Elks, P.M.; Loynes, C.A.; Whyte, M.K.; Renshaw, S.A. A method for the in vivo measurement of zebrafish tissue neutrophil lifespan. ISRN Hematol. 2012, 2012, 915868. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Skubitz, K.; Micklem, K.; van der Schoot, E. CD66 and CD67 cluster workshop report. In Leukocyte Typing V; Schlossmann, S.F., Boumsell, L., Gilks, W., Harlan, J., Kishimoto, T., Morimoto, C., Ritz, J., Springer, T.A., Tedder, T.F., Todd, R.F., Eds.; Oxford University Press: Oxford, UK, 1995; Volume 1, pp. 889–899. [Google Scholar]
- Bao, Y.; Cao, X. Revisiting the protective and pathogenic roles of neutrophils: Ly-6G is key! Eur. J. Immunol. 2011, 41, 2535–2538. [Google Scholar] [CrossRef]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K.; et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Massena, S.; Christoffersson, G.; Vagesjo, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Herrera, H.C.; Giraud, A.; Lomei, J.; Westrom, S.; et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29, 1334–1342. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [Green Version]
- Lecot, P.; Sarabi, M.; Pereira, A.M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Sherer, Y.; Gorstein, A.; Fritzler, M.J.; Shoenfeld, Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Semin. Arthritis Rheum. 2004, 34, 501–537. [Google Scholar] [CrossRef]
- Rumore, P.M.; Steinman, C.R. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Investig. 1990, 86, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Amoura, Z.; Piette, J.C.; Chabre, H.; Cacoub, P.; Papo, T.; Wechsler, B.; Bach, J.F.; Koutouzov, S. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus: Correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum. 1997, 40, 2217–2225. [Google Scholar] [CrossRef]
- Williams, R.C., Jr.; Malone, C.C.; Meyers, C.; Decker, P.; Muller, S. Detection of nucleosome particles in serum and plasma from patients with systemic lupus erythematosus using monoclonal antibody 4H7. J. Rheumatol. 2001, 28, 81–94. [Google Scholar]
- Amoura, Z.; Koutouzov, S.; Chabre, H.; Cacoub, P.; Amoura, I.; Musset, L.; Bach, J.F.; Piette, J.C. Presence of antinucleosome autoantibodies in a restricted set of connective tissue diseases: Antinucleosome antibodies of the IgG3 subclass are markers of renal pathogenicity in systemic lupus erythematosus. Arthritis Rheum. 2000, 43, 76–84. [Google Scholar] [CrossRef]
- Lu, L.; Kaliyaperumal, A.; Boumpas, D.T.; Datta, S.K. Major peptide autoepitopes for nucleosome-specific T cells of human lupus. J. Clin. Investig. 1999, 104, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Schellekens, G.A.; de Jong, B.A.; van den Hoogen, F.H.; van de Putte, L.B.; van Venrooij, W.J. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Girbal-Neuhauser, E.; Durieux, J.J.; Arnaud, M.; Dalbon, P.; Sebbag, M.; Vincent, C.; Simon, M.; Senshu, T.; Masson-Bessiere, C.; Jolivet-Reynaud, C.; et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J. Immunol. 1999, 162, 585–594. [Google Scholar]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooij, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Sharma, S.; Davis, R.E.; Srivastva, S.; Nylen, S.; Sundar, S.; Wilson, M.E. A Subset of Neutrophils Expressing Markers of Antigen-Presenting Cells in Human Visceral Leishmaniasis. J. Infect. Dis. 2016, 214, 1531–1538. [Google Scholar] [CrossRef]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Beauvillain, C.; Delneste, Y.; Scotet, M.; Peres, A.; Gascan, H.; Guermonprez, P.; Barnaba, V.; Jeannin, P. Neutrophils efficiently cross-prime naive T cells in vivo. Blood 2007, 110, 2965–2973. [Google Scholar] [CrossRef]
- Davey, M.S.; Morgan, M.P.; Liuzzi, A.R.; Tyler, C.J.; Khan, M.W.A.; Szakmany, T.; Hall, J.E.; Moser, B.; Eberl, M. Microbe-specific unconventional T cells induce human neutrophil differentiation into antigen cross-presenting cells. J. Immunol. 2014, 193, 3704–3716. [Google Scholar] [CrossRef] [Green Version]
- Lindau, D.; Mussard, J.; Wagner, B.J.; Ribon, M.; Ronnefarth, V.M.; Quettier, M.; Jelcic, I.; Boissier, M.C.; Rammensee, H.G.; Decker, P. Primary blood neutrophils express a functional cell surface Toll-like receptor 9. Eur. J. Immunol. 2013, 43, 2101–2113. [Google Scholar] [CrossRef]
- Lindau, D.; Mussard, J.; Rabsteyn, A.; Ribon, M.; Kotter, I.; Igney, A.; Adema, G.J.; Boissier, M.C.; Rammensee, H.G.; Decker, P. TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann. Rheum. Dis. 2014, 73, 2199–2207. [Google Scholar] [CrossRef]
- Puellmann, K.; Kaminski, W.E.; Vogel, M.; Nebe, C.T.; Schroeder, J.; Wolf, H.; Beham, A.W. A variable immunoreceptor in a subpopulation of human neutrophils. Proc. Natl. Acad. Sci. USA 2006, 103, 14441–14446. [Google Scholar] [CrossRef] [Green Version]
- Costantini, C.; Calzetti, F.; Perbellini, O.; Micheletti, A.; Scarponi, C.; Lonardi, S.; Pelletier, M.; Schakel, K.; Pizzolo, G.; Facchetti, F.; et al. Human neutrophils interact with both 6-sulfo LacNAc+ DC and NK cells to amplify NK-derived IFNγ: Role of CD18, ICAM-1, and ICAM-3. Blood 2011, 117, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2012, 13, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Sadatomo, A.; Inoue, Y.; Ito, H.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mizushina, Y.; Nakamura, J.; Kamata, R.; Kasahara, T.; et al. Interaction of Neutrophils with Macrophages Promotes IL-1β Maturation and Contributes to Hepatic Ischemia-Reperfusion Injury. J. Immunol. 2017, 199, 3306–3315. [Google Scholar] [CrossRef] [Green Version]
- Wantha, S.; Alard, J.E.; Megens, R.T.; van der Does, A.M.; Doring, Y.; Drechsler, M.; Pham, C.T.; Wang, M.W.; Wang, J.M.; Gallo, R.L.; et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ. Res. 2013, 112, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Batignes, M.; Santinon, F.; Boissier, M.C.; Decker, P.; Bessis, N. Neutrophils and Regulatory T Lymphocytes (Treg) Cooperate to Sustain Treg Activity but This Interaction Is Altered in Rheumatoid Arthritis Patients [abstract]. Arthritis Rheumatol. 2019, 71, 163–164. [Google Scholar]
- Piskin, G.; Bos, J.D.; Teunissen, M.B. Neutrophils infiltrating ultraviolet B-irradiated normal human skin display high IL-10 expression. Arch. Dermatol. Res. 2005, 296, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Ribon, M.; Mussard, J.; Semerano, L.; Singer, B.B.; Decker, P. Extracellular Chromatin Triggers Release of Soluble CEACAM8 Upon Activation of Neutrophils. Front. Immunol. 2019, 10, 1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, V.M.; Pillay, J.; Lammers, J.W.; Pickkers, P.; Ulfman, L.H.; Koenderman, L. Human suppressive neutrophils CD16bright/CD62Ldim exhibit decreased adhesion. J. Leukoc. Biol. 2012, 92, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T.; et al. Mature CD10+ and immature CD10− neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood 2017, 129, 1343–1356. [Google Scholar] [CrossRef] [Green Version]
- Wingender, G.; Hiss, M.; Engel, I.; Peukert, K.; Ley, K.; Haller, H.; Kronenberg, M.; von Vietinghoff, S. Neutrophilic granulocytes modulate invariant NKT cell function in mice and humans. J. Immunol. 2012, 188, 3000–3008. [Google Scholar] [CrossRef] [Green Version]
- Gresnigt, M.S.; Joosten, L.A.; Verschueren, I.; van der Meer, J.W.; Netea, M.G.; Dinarello, C.A.; van de Veerdonk, F.L. Neutrophil-mediated inhibition of proinflammatory cytokine responses. J. Immunol. 2012, 189, 4806–4815. [Google Scholar] [CrossRef] [Green Version]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef]
- Barrientos, L.; Bignon, A.; Gueguen, C.; de Chaisemartin, L.; Gorges, R.; Sandre, C.; Mascarell, L.; Balabanian, K.; Kerdine-Romer, S.; Pallardy, M.; et al. Neutrophil extracellular traps downregulate lipopolysaccharide-induced activation of monocyte-derived dendritic cells. J. Immunol. 2014, 193, 5689–5698. [Google Scholar] [CrossRef] [Green Version]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Gestermann, N.; Di Domizio, J.; Lande, R.; Demaria, O.; Frasca, L.; Feldmeyer, L.; Di Lucca, J.; Gilliet, M. Netting Neutrophils Activate Autoreactive B Cells in Lupus. J. Immunol. 2018, 200, 3364–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET: Current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Abi Abdallah, D.S.; Lin, C.; Ball, C.J.; King, M.R.; Duhamel, G.E.; Denkers, E.Y. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect. Immun. 2012, 80, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 423. [Google Scholar] [CrossRef]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar]
- Lauth, X.; Kockritz-Blickwede, M.; McNamara, C.W.; Myskowski, S.; Zinkernagel, A.S.; Beall, B.; Ghosh, P.; Gallo, R.L.; Nizet, V. M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J. Innate Immun. 2009, 1, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [Green Version]
- Derré-Bobillot, A.; Cortes-Perez, N.G.; Yamamoto, Y.; Kharrat, P.; Couvé, E.; Da Cunha, V.; Decker, P.; Boissier, M.C.; Escartin, F.; Cesselin, B.; et al. Nuclease A (Gbs0661), an extracellular nuclease of Streptococcus agalactiae, attacks the neutrophil extracellular traps and is needed for full virulence. Mol. Microbiol. 2013, 89, 518–531. [Google Scholar] [CrossRef]
- Neumann, A.; Vollger, L.; Berends, E.T.; Molhoek, E.M.; Stapels, D.A.; Midon, M.; Friaes, A.; Pingoud, A.; Rooijakkers, S.H.; Gallo, R.L.; et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. J. Innate Immun. 2014, 6, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T.; et al. Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe 2019, 25, 526–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Parker, H.; Dragunow, M.; Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 2012, 92, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.F.; Herzig, A.; Kruger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Bernuth, H.V.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6, e24437. [Google Scholar] [CrossRef]
- DeSouza-Vieira, T.; Guimaraes-Costa, A.; Rochael, N.C.; Lira, M.N.; Nascimento, M.T.; Lima-Gomez, P.S.; Mariante, R.M.; Persechini, P.M.; Saraiva, E.M. Neutrophil extracellular traps release induced by Leishmania: Role of PI3Kgamma, ERK, PI3Ksigma, PKC, and [Ca2+]. J. Leukoc. Biol. 2016, 100, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Kenno, S.; Perito, S.; Mosci, P.; Vecchiarelli, A.; Monari, C. Autophagy and Reactive Oxygen Species Are Involved in Neutrophil Extracellular Traps Release Induced by C. albicans Morphotypes. Front. Microbiol. 2016, 7, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Behnen, M.; Leschczyk, C.; Moller, S.; Batel, T.; Klinger, M.; Solbach, W.; Laskay, T. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac-1. J. Immunol. 2014, 193, 1954–1965. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Wang, X.; Wicki, S.; Kaufmann, T.; Wong, W.W.; Simon, H.U.; Yousefi, S. NET formation can occur independently of RIPK3 and MLKL signaling. Eur. J. Immunol. 2016, 46, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 2958. [Google Scholar] [CrossRef] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [Green Version]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Padrines, M.; Wolf, M.; Walz, A.; Baggiolini, M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994, 352, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Molad, Y.; Buyon, J.; Anderson, D.C.; Abramson, S.B.; Cronstein, B.N. Intravascular neutrophil activation in systemic lupus erythematosus (SLE): Dissociation between increased expression of CD11b/CD18 and diminished expression of L-selectin on neutrophils from patients with active SLE. Clin. Immunol. Immunopathol. 1994, 71, 281–286. [Google Scholar] [CrossRef]
- Wright, H.L.; Chikura, B.; Bucknall, R.C.; Moots, R.J.; Edwards, S.W. Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann. Rheum. Dis. 2011, 70, 537–543. [Google Scholar] [CrossRef]
- Wright, H.L.; Moots, R.J.; Edwards, S.W. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Rumore, P.M.; Liu, Q.; Steinman, C.R. Soluble oligonucleosomal complexes in synovial fluid from inflamed joints. Arthritis Rheum. 1997, 40, 648–654. [Google Scholar] [CrossRef]
- Mohr, W.; Pelster, B.; Wessinghage, D. Polymorphonuclear granulocytes in rheumatic tissue destruction. VI. The occurrence of PMNs in menisci of patients with rheumatoid arthritis. Rheumatol. Int. 1984, 5, 39–44. [Google Scholar] [CrossRef]
- De Bont, C.M.; Stokman, M.E.M.; Faas, P.; Thurlings, R.M.; Boelens, W.C.; Wright, H.L.; Pruijn, G.J.M. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J. Autoimmun. 2020, 113, 102484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, J.; Kucik, D.F.; White, N.B.; Redden, D.T.; Szalai, A.J.; Bullard, D.C.; Edberg, J.C. Multiple lupus-associated ITGAM variants alter Mac-1 functions on neutrophils. Arthritis Rheum. 2013, 65, 2907–2916. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Arve, S.; Ledbetter, J.; Elkon, K.B. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J. Exp. Med. 2017, 214, 2103–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Huang, Z.; Ye, J.; Deng, Y.; Fang, L.; Li, X.; Guo, Y.; Jiang, H.; Ju, B.; Huang, Q.; et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, P.; Rodriguez-Carrio, J.; Caminal-Montero, L.; Mozo, L.; Suarez, A. A pathogenic IFNalpha, BLyS and IL-17 axis in Systemic Lupus Erythematosus patients. Sci. Rep. 2016, 6, 20651. [Google Scholar] [CrossRef]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef]
- Rahman, S.; Sagar, D.; Hanna, R.N.; Lightfoot, Y.L.; Mistry, P.; Smith, C.K.; Manna, Z.; Hasni, S.; Siegel, R.M.; Sanjuan, M.A.; et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 957–966. [Google Scholar] [CrossRef] [Green Version]
- Van Bruggen, M.C.; Kramers, C.; Walgreen, B.; Elema, J.D.; Kallenberg, C.G.; van den Born, J.; Smeenk, R.J.; Assmann, K.J.; Muller, S.; Monestier, M.; et al. Nucleosomes and histones are present in glomerular deposits in human lupus nephritis. Nephrol. Dial. Transplant. 1997, 12, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Rönnefarth, V.M.; Erbacher, A.I.; Lamkemeyer, T.; Madlung, J.; Nordheim, A.; Rammensee, H.G.; Decker, P. TLR2/TLR4-independent neutrophil activation and recruitment upon endocytosis of nucleosomes reveals a new pathway of innate immunity in systemic lupus erythematosus. J. Immunol. 2006, 177, 7740–7749. [Google Scholar] [CrossRef] [Green Version]
- Lindau, D.; Rönnefarth, V.; Erbacher, A.; Rammensee, H.G.; Decker, P. Nucleosome-induced neutrophil activation occurs independently of TLR9 and endosomal acidification: Implications for systemic lupus erythematosus. Eur. J. Immunol. 2011, 41, 669–681. [Google Scholar] [CrossRef]
- Palanichamy, A.; Bauer, J.W.; Yalavarthi, S.; Meednu, N.; Barnard, J.; Owen, T.; Cistrone, C.; Bird, A.; Rabinovich, A.; Nevarez, S.; et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J. Immunol. 2014, 192, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Hervier, B.; Ribon, M.; Tarantino, N.; Mussard, J.; Breckler, M.; Vieillard, V.; Amoura, Z.; Steinle, A.; Klein, R.; Kotter, I.; et al. Increased Concentrations of Circulating Soluble MHC Class I-Related Chain A (sMICA) and sMICB and Modulation of Plasma Membrane MICA Expression: Potential Mechanisms and Correlation With Natural Killer Cell Activity in Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 633658. [Google Scholar] [CrossRef] [PubMed]
- Means, T.K.; Latz, E.; Hayashi, F.; Murali, M.R.; Golenbock, D.T.; Luster, A.D. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Investig. 2005, 115, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Patino-Trives, A.M.; Perez-Sanchez, C.; Perez-Sanchez, L.; Luque-Tevar, M.; Abalos-Aguilera, M.C.; Alcaide-Ruggiero, L.; Arias-de la Rosa, I.; Roman-Rodriguez, C.; Segui, P.; Espinosa, M.; et al. Anti-dsDNA Antibodies Increase the Cardiovascular Risk in Systemic Lupus Erythematosus Promoting a Distinctive Immune and Vascular Activation. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2417–2430. [Google Scholar] [CrossRef]
- Chitrabamrung, S.; Rubin, R.L.; Tan, E.M. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol. Int. 1981, 1, 55–60. [Google Scholar] [CrossRef]
- Al Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Bijl, M.; Reefman, E.; Horst, G.; Limburg, P.C.; Kallenberg, C.G. Reduced uptake of apoptotic cells by macrophages in Systemic Lupus Erythematosus (SLE): Correlates with decreased serum levels of complement. Ann. Rheum. Dis. 2006, 65, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Barrera-Vargas, A.; Gomez-Martin, D.; Carmona-Rivera, C.; Merayo-Chalico, J.; Torres-Ruiz, J.; Manna, Z.; Hasni, S.; Alcocer-Varela, J.; Kaplan, M.J. Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 944–950. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.; Petretto, A.; Santucci, L.; Vaglio, A.; Pratesi, F.; Migliorini, P.; Bertelli, R.; Lavarello, C.; Bartolucci, M.; Candiano, G.; et al. Neutrophil Extracellular Traps protein composition is specific for patients with Lupus nephritis and includes methyl-oxidized alphaenolase (methionine sulfoxide 93). Sci. Rep. 2019, 9, 7934. [Google Scholar] [CrossRef]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Georgakis, S.; Gkirtzimanaki, K.; Papadaki, G.; Gakiopoulou, H.; Drakos, E.; Eloranta, M.L.; Makridakis, M.; Kontostathi, G.; Zoidakis, J.; Baira, E.; et al. NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFNalpha production in SLE patients. JCI Insight 2021, 6, e147671. [Google Scholar]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; Van Der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Wojciak-Stothard, B.; Ruseva, M.M.; Cook, H.T.; Kelleher, P.; Pickering, M.C.; Mongkolsapaya, J.; Screaton, G.R.; Xu, X.N. Autoantibody-dependent amplification of inflammation in SLE. Cell Death Dis. 2020, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Kaplan, M.J. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 2012, 24, 441–450. [Google Scholar] [CrossRef]
- Knight, J.S.; Carmona-Rivera, C.; Kaplan, M.J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 2012, 3, 380. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Rauova, L.; Gilburd, B.; Zurgil, N.; Blank, M.; Guegas, L.L.; Brickman, C.M.; Cebecauer, L.; Deutsch, M.; Wiik, A.; Shoenfeld, Y. Induction of biologically active antineutrophil cytoplasmic antibodies by immunization with human apoptotic polymorphonuclear leukocytes. Clin. Immunol. 2002, 103, 69–78. [Google Scholar] [CrossRef]
- Mourao, A.F.; Canhao, H.; Sousa, E.; Cascao, R.; da Costa, J.B.; de Almeida, L.S.; Oliveira, M.E.; Gomes, M.M.; Queiroz, M.V.; Fonseca, J.E. From a neutrophilic synovial tissue infiltrate to a challenging case of rheumatoid arthritis. Acta Reumatol. Port. 2010, 35, 228–231. [Google Scholar] [PubMed]
- Cross, A.; Bucknall, R.C.; Cassatella, M.A.; Edwards, S.W.; Moots, R.J. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2796–2806. [Google Scholar] [CrossRef]
- Iking-Konert, C.; Ostendorf, B.; Sander, O.; Jost, M.; Wagner, C.; Joosten, L.; Schneider, M.; Hansch, G.M. Transdifferentiation of polymorphonuclear neutrophils to dendritic-like cells at the site of inflammation in rheumatoid arthritis: Evidence for activation by T cells. Ann. Rheum. Dis. 2005, 64, 1436–1442. [Google Scholar] [CrossRef] [Green Version]
- Cross, A.; Barnes, T.; Bucknall, R.C.; Edwards, S.W.; Moots, R.J. Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J. Leukoc. Biol. 2006, 80, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Yeo, L.; Toellner, K.M.; Salmon, M.; Filer, A.; Buckley, C.D.; Raza, K.; Scheel-Toellner, D. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 2022–2028. [Google Scholar] [CrossRef] [Green Version]
- Assi, L.K.; Wong, S.H.; Ludwig, A.; Raza, K.; Gordon, C.; Salmon, M.; Lord, J.M.; Scheel-Toellner, D. Tumor necrosis factor alpha activates release of B lymphocyte stimulator by neutrophils infiltrating the rheumatoid joint. Arthritis Rheum. 2007, 56, 1776–1786. [Google Scholar] [CrossRef] [Green Version]
- Contis, A.; Mitrovic, S.; Lavie, J.; Douchet, I.; Lazaro, E.; Truchetet, M.E.; Goizet, C.; Contin-Bordes, C.; Schaeverbeke, T.; Blanco, P.; et al. Neutrophil-derived mitochondrial DNA promotes receptor activator of nuclear factor kappaB and its ligand signalling in rheumatoid arthritis. Rheumatology 2017, 56, 1200–1205. [Google Scholar] [CrossRef] [Green Version]
- Poubelle, P.E.; Chakravarti, A.; Fernandes, M.J.; Doiron, K.; Marceau, A.A. Differential expression of RANK, RANK-L, and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Res. Ther. 2007, 9, R25. [Google Scholar] [CrossRef] [Green Version]
- Wright, H.L.; Lyon, M.; Chapman, E.A.; Moots, R.J.; Edwards, S.W. Rheumatoid Arthritis Synovial Fluid Neutrophils Drive Inflammation Through Production of Chemokines, Reactive Oxygen Species, and Neutrophil Extracellular Traps. Front. Immunol. 2020, 11, 584116. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Cox, T.; Moots, R.J.; Edwards, S.W. Neutrophil biomarkers predict response to therapy with tumor necrosis factor inhibitors in rheumatoid arthritis. J. Leukoc. Biol. 2017, 101, 785–795. [Google Scholar] [CrossRef]
- Wright, H.L.; Makki, F.A.; Moots, R.J.; Edwards, S.W. Low-density granulocytes: Functionally distinct, immature neutrophils in rheumatoid arthritis with altered properties and defective TNF signalling. J. Leukoc. Biol. 2017, 101, 599–611. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [Green Version]
- Papadaki, G.; Kambas, K.; Choulaki, C.; Vlachou, K.; Drakos, E.; Bertsias, G.; Ritis, K.; Boumpas, D.T.; Thompson, P.R.; Verginis, P.; et al. Neutrophil extracellular traps exacerbate Th1-mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur. J. Immunol. 2016, 46, 2542–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar]
- Carmona-Rivera, C.; Carlucci, P.M.; Moore, E.; Lingampalli, N.; Uchtenhagen, H.; James, E.; Liu, Y.; Bicker, K.; Wahamaa, H.; Hoffmann, V.; et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci. Immunol. 2017, 2, eaag3358. [Google Scholar]
- Pratesi, F.; Dioni, I.; Tommasi, C.; Alcaro, M.C.; Paolini, I.; Barbetti, F.; Boscaro, F.; Panza, F.; Puxeddu, I.; Rovero, P.; et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann. Rheum. Dis. 2014, 73, 1414–1422. [Google Scholar]
- Corsiero, E.; Bombardieri, M.; Carlotti, E.; Pratesi, F.; Robinson, W.; Migliorini, P.; Pitzalis, C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann. Rheum. Dis. 2016, 75, 1866–1875. [Google Scholar]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.; van Veelen, P.A.; Levarht, N.E.; van der Helm-van Mil, A.H.; Cerami, A.; Huizinga, T.W.; et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar]
- O’Neil, L.J.; Barrera-Vargas, A.; Sandoval-Heglund, D.; Merayo-Chalico, J.; Aguirre-Aguilar, E.; Aponte, A.M.; Ruiz-Perdomo, Y.; Gucek, M.; El Gabalawy, H.; Fox, D.A.; et al. Neutrophil-mediated carbamylation promotes articular damage in rheumatoid arthritis. Sci. Adv. 2020, 6, eabd2688. [Google Scholar] [CrossRef]
- Mustila, A.; Paimela, L.; Leirisalo-Repo, M.; Huhtala, H.; Miettinen, A. Antineutrophil cytoplasmic antibodies in patients with early rheumatoid arthritis: An early marker of progressive erosive disease. Arthritis Rheum. 2000, 43, 1371–1377. [Google Scholar]
- Hu, L.; Hu, X.; Long, K.; Gao, C.; Dong, H.L.; Zhong, Q.; Gao, X.M.; Gong, F.Y. Extraordinarily potent proinflammatory properties of lactoferrin-containing immunocomplexes against human monocytes and macrophages. Sci. Rep. 2017, 7, 4230. [Google Scholar]
- Fossati, G.; Bucknall, R.C.; Edwards, S.W. Insoluble and soluble immune complexes activate neutrophils by distinct activation mechanisms: Changes in functional responses induced by priming with cytokines. Ann. Rheum. Dis. 2002, 61, 13–19. [Google Scholar]
- Robinson, J.J.; Watson, F.; Bucknall, R.C.; Edwards, S.W. Role of Fc gamma receptors in the activation of neutrophils by soluble and insoluble immunoglobulin aggregates isolated from the synovial fluid of patients with rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 515–520. [Google Scholar]
- Carmona-Rivera, C.; Carlucci, P.M.; Goel, R.R.; James, E.; Brooks, S.R.; Rims, C.; Hoffmann, V.; Fox, D.A.; Buckner, J.H.; Kaplan, M.J. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight 2020, 5, e139388. [Google Scholar] [CrossRef] [PubMed]
- Monach, P.A.; Hueber, W.; Kessler, B.; Tomooka, B.H.; BenBarak, M.; Simmons, B.P.; Wright, J.; Thornhill, T.S.; Monestier, M.; Ploegh, H.; et al. A broad screen for targets of immune complexes decorating arthritic joints highlights deposition of nucleosomes in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 15867–15872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.; Moon, J.; Moore, R.; Pan, T.; Nelson, J.L.; Lood, C. A Neutrophil Activation Biomarker Panel in Prognosis and Monitoring of Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2020, 72, 47–56. [Google Scholar] [CrossRef]
- Perez-Sanchez, C.; Ruiz-Limon, P.; Aguirre, M.A.; Jimenez-Gomez, Y.; Arias-de la Rosa, I.; Abalos-Aguilera, M.C.; Rodriguez-Ariza, A.; Castro-Villegas, M.C.; Ortega-Castro, R.; Segui, P.; et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in Rheumatoid Arthritis patients. J. Autoimmun. 2017, 82, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yang, C.; Wang, J.; Li, Y.; Yang, P. Serum level of DNase1l3 in patients with dermatomyositis/polymyositis, systemic lupus erythematosus and rheumatoid arthritis, and its association with disease activity. Clin. Exp. Med. 2017, 17, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Martinez-Bonet, M.; Onengut-Gumuscu, S.; Lee, A.; Luo, Y.; Teslovich, N.; Worthington, J.; Martin, J.; Huizinga, T.; Klareskog, L.; et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018, 50, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.U.; et al. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef] [Green Version]
- von Kockritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Granger, V.; Faille, D.; Marani, V.; Noel, B.; Gallais, Y.; Szely, N.; Flament, H.; Pallardy, M.; Chollet-Martin, S.; de Chaisemartin, L. Human blood monocytes are able to form extracellular traps. J. Leukoc. Biol. 2017, 102, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelsson, B.; Soderberg, D.; Strid, T.; Soderberg, A.; Bergh, A.C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosen, A. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haidar Ahmad, A.; Melbouci, D.; Decker, P. Polymorphonuclear Neutrophils in Rheumatoid Arthritis and Systemic Lupus Erythematosus: More Complicated Than Anticipated. Immuno 2022, 2, 85-103. https://doi.org/10.3390/immuno2010007
Haidar Ahmad A, Melbouci D, Decker P. Polymorphonuclear Neutrophils in Rheumatoid Arthritis and Systemic Lupus Erythematosus: More Complicated Than Anticipated. Immuno. 2022; 2(1):85-103. https://doi.org/10.3390/immuno2010007
Chicago/Turabian StyleHaidar Ahmad, Ahmad, Dyhia Melbouci, and Patrice Decker. 2022. "Polymorphonuclear Neutrophils in Rheumatoid Arthritis and Systemic Lupus Erythematosus: More Complicated Than Anticipated" Immuno 2, no. 1: 85-103. https://doi.org/10.3390/immuno2010007