1. Introduction
The NLRP3 inflammasome is a multiprotein complex that forms in the cytosol, which, when assembled, activates caspase-1 to cleave the proinflammatory cytokine interleukin (IL)-1β [
1]. Inflammasome activation requires two signals: first, the transcription and translation of NLRP3 and pro IL-1β, and second, the formation of the NLRP3 multiprotein complex, driving cleavage of caspase-1 and leading to the maturation and secretion of mature IL-1β. The inflammasome has a role in several chronic, immune-mediated, mucosal illnesses. These include the inflammatory bowel diseases (IBD), including Crohn disease (CD) and ulcerative colitis (UC), which are chronic intestinal illnesses with increasing incidence [
2]. The etiology of IBD remains unknown, but there is evidence suggesting a link between genes, the environment, and microbes, leading to an uncontrolled proinflammatory response [
3,
4,
5,
6]. Although over 200 genetic loci are linked to increased susceptibility to IBD, no single gene carries a high enough weight to determine the exact cause. Interestingly, the
NLRP3 gene, which plays a major role in bacterial recognition and immune responses in immune cells, has been linked to IBD. Furthermore, and in contrast to its role in sterile inflammatory conditions, such as gout or rheumatoid arthritis, where NLRP3 has been shown to be involved in disease etiology [
7], NLRP3 may play a protective role in IBD, specifically in preventing colitis [
8].
One model commonly used to study NLRP3 and IBD utilizes
Citrobacter rodentium, a Gram-negative bacterium that causes acute colitis in mice and has been used extensively as a model of infection-induced inflammation, similar in some ways to that seen in IBD [
9,
10]. We have previously shown that
NLRP3−/− mice infected with
C. rodentium display higher colonization rates and that this can be alleviated by injecting exogenous IL-1β [
9]. In another study, we used
C. rodentium and extracellular ATP to induce inflammasome activation in macrophages where the bacterium can activate both signals to promote inflammasome activity, while extracellular ATP triggers only signal two through the activation of the purinergic receptor P
2X7 [
11]. We showed that macrophages exposed to extracellular ATP during infection with
C. rodentium display increased bacterial killing, along with reduced proinflammatory responses. The purinergic receptor P
2X7 is ubiquitously expressed throughout all human cells, with unique pathways in each cell type [
12]. Furthermore, P
2X7 activation in gut epithelial cells during infection induces expression of CCL-5, a chemokine that drives recruitment of macrophages [
13].
The intestinal epithelial layer forms a physical barrier between the microbe-rich environment of the lumen and the mucosal immune system, further aiding in preventing infection within the gut [
14]. Homeostasis of this environment is maintained by the integrity of the intestinal epithelial cell monolayer and is critical for gastrointestinal health. Epithelial cells have innate immune functions where they can detect potential pathogens through innate recognition, secrete cytokines and chemokine for immune activation, and provide defense through secretion of antimicrobial peptides (AMP) into the intestinal tract [
15]. Inflammasomes are not well characterized in intestinal epithelial cells; however, it is known that NLRP6 is required for mucous secretion from goblet cells, a function depleted in IBD [
4]. Yet, there is limited knowledge on the function of the NLRP3 inflammasome in intestinal epithelial cells, as NLRP3 has been mostly characterized in macrophages. A recent publication has shown that during
Toxoplasma gondii infection, small intestinal expression of
NLRP3, through the purinergic receptor P
2X7 and subsequent IL-1β secretion, has a protective effect [
16].
We aimed to study the role of the inflammasome in murine intestinal epithelial cells during infection with C. rodentium and how this is affected by macrophages. The complex epithelial–macrophage relationship is critical for innate immunity within the intestinal tract during infection and maintenance of the host microbiome. An in vitro co-culture model allowed us to analyze the role of the inflammasome in this complex relationship. We initially hypothesized that activation of the NLRP3 inflammasome in epithelial cells would improve their recovery after infection with C. rodentium. Interestingly, we found that this was not the case, as ATP-activation of the inflammasome in intestinal epithelial cells did not directly enhance barrier recovery; however, secreted factors from epithelial cells decreased bacterial survival inside macrophages (likely by improving bacterial clearance). We also found that ATP-activated macrophages aided in recovery of epithelial barrier after infection, that macrophages were recruited to the apical epithelial membrane, and that expression of tight junction proteins was altered in macrophage cells during infection. Although the study of inflammasome activation may be currently focused on macrophages, epithelial cells play an important role in innate immunity, through interactions with macrophages, and understanding the role of the inflammasome in these cells is of great importance.
2. Materials and Methods
2.1. Cell Culture
J774A.1 murine macrophage cell line (ATCC, TIB-67) and CMT-93 murine colonic cell line (ATCC, CCL-223) were seeded as indicated for each experiment. Cells were maintained in DMEM (Life Technologies, Burlington, ON, Canada), supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies); the medium was replaced every two days and cells were passaged at 80% confluence for a maximum of 22 passages from thaw.
2.2. C. rodentium Infection
CMT-93 or J774A.1 cells were seeded in 24-well tissue culture plates at a density of 5 × 105 cells per well (~80% confluence) overnight to allow for adhesion. Prior to infection, the medium was changed to antibiotic and serum-free DMEM. C. rodentium (DBS100, a gift from the Sherman lab, University of Toronto, Toronto, ON, Canada) was cultured overnight in lysogeny broth (LB) at 37 °C. Cells were treated with the caspase 1 inhibitor AC-YVAD-CMK (YVAD: 25 μM, SML0429; Enzo Life Sciences, Sigma-Aldritch, Oakville, ON, Canada) to inhibit the inflammasome for 1 h pre-infection.
C. rodentium was added at a multiplicity of infection (MOI) of 10:1 for 4 h of infection; ATP (2.5 mM) was then added for 30 min. Supernatants were collected, with the addition of protease inhibitor cocktail (1:100 dilution, P8340, Sigma-Aldritch, Oakville, ON, Canada), for IL-18 and IL-1β secretion measured using ELISA (DY7625-5 and DY201-5, R & D systems). Cells were washed thrice with 1× PBS, lysed with Triton X-100 (1%, Thermo Fisher, Mississauga, ON, Canada), and plated on LB agar overnight at 37 °C after serial dilutions to enumerate adhesive bacteria. Bacterial adherence to epithelial cells on the Transwell was assessed by adding Triton X-100 after the 24-h infection and plating on LB agar overnight; results are expressed as a percentage, compared to control, without the presence of macrophages.
Supernatants from infected epithelial cells (with or without inflammasome activation/inhibition) were collected after 24-h infection, centrifuged (7500 rpm, 5 min) and then syringe-filtered (2 µm). Supernatants were 10× diluted and then added to macrophages to assess the effects of factors secreted by epithelial cells on microbial killing by macrophages.
2.3. Epithelial Layer Barrier Function Assessment Using Transepithelial Electrical Resistance (TEER)
In order to best recreate the gut epithelial-macrophage-microbial interface in an in vitro setting, the following model was developed. The underside of Transwells (3 µm pore size, Corning) were coated with rat tail collagen (0.33 mg/mL, Gibco) and then seeded with 1 × 10
5 cells per mL of CMT-93 murine colonic cells until confluence, measured using TEER, shown as a plateau in resistance. Before each measurement, the chopsticks were sterilized using ethanol and then washed once with 1× PBS. After achieving confluence, the Transwell inserts were flipped into a 24 well petri dish with 5 mL of PBS. 50 µL of GFP-
C. rodentium from overnight broth (approximately 5 × 10
6 CFU/mL) was resuspended in DMEM and then added to the apical membrane for 3 h. After the 3 h infection, the inserts were washed once with 1X PBS to remove any not adhered bacteria and added to a new 24-well plate, in the standard orientation (apical side up) (
Supplemental Figure S1). ATP (2.5 mM, BP413-25; Fisher Scientific, Waltham, MA, USA), YVAD (25 µM), or both were added to the basolateral membrane, where indicated. Treated and non-treated J774A.1 macrophages were then added to the basolateral chamber and Gentamicin (10 µg/mL, Sigma-Aldritch, Oakville, Canada) was added to the apical side to prevent bacterial overgrowth (does not penetrate eukaryotic cell membranes). Supernatants were collected as described above for IL-1β secretion. Supernatants of infected J774A.1 macrophages with and without ATP were filtered (0.2 µm pore) and added to the basolateral chamber, where indicated (10× dilution), to assess the effects of factors secreted from epithelial cells. TEER was then measured over a 24-h period with results presented as percent recovery, calculated relative to the initial resistance measurement (in ohm) just before infection. The resistance of a blank insert without cells was measured in parallel and subtracted from the sample resistance to reflect the natural resistance of the Transwell and fluid.
2.4. Immunofluorescence of Actin, Macrophages, and Tight Junction ZO-1
Transwells were fixed with either methanol (for ZO-1 staining) or 4% PFA (all other stainings), and then blocked in 2% Goat Serum and 1% bovine serum albumin (15 min). Actin was stained with Alexa Fluor-594 phalloidin (1:40 dilution, 0.1% Triton, 0.2% Goat Serum, and 0.1% BSA; Thermo Fisher, Mississauga, Canada); 4′,6-diamidino-2-phenylindole (DAPI) (1:1000 dilution, 0.1% Triton, 0.2% Goat Serum, and 0.1% BSA; Thermo Fisher) was used for nuclear staining. Additional stains included zone occluding (ZO)-1 (1:200 dilution, 0.1% Triton, 0.2% Goat Serum, and 0.1% BSA, Abcam, Toronto, Canada), F4/80 (murine macrophage marker; 1:200; Thermo Fisher, Mississauga, Canada), and green fluorescent protein
C. rodentium (GFP-DBS 100; a generous gift from Dr. Bruce Vallance, University of British Columbia, Vancouver, Canada). Transwells were cut out and mounted on slides, which were analyzed using the Zeiss Axio Observer.Z1 microscope with ZEN Imaging software (Carl Zeiss Canada Ltd., Toronto, ON, Canada); illustrations were identically formatted for noise reduction and increased sharpness using FIJI (National Institutes of Health, Bethesda, Rockville, MD, USA) [
17].
2.5. qPCR for Tight Junction and NLRP3 Genes
J774A.1 and CMT-93 cells were cultured overnight prior to treatment as indicated. RNA was isolated using 1 mL Trizol (Invitrogen), as previously described [
18]. RT-qPCR was performed as previously described to validate findings, using primers highlighted in
Table 1 [
18]. Both biological and technical replicates were performed on all reactions using
Rer1,
Rpl 21, and
Rpl 27 as housekeeping genes. Data were analyzed using CFX Manager Software Version 3.0 (Bio-Rad Laboratories, Inc., Mississauga, Canada). Statistical significance was evaluated by one-way ANOVA using GraphPad Prism 9.0 (San Diego, CA, USA).
2.6. Western Blot for Tight Junction Proteins
J774A.1 cells were seeded in 10 cm tissue culture plates until confluent and infected as stated above. Cells were treated with the caspase 1 inhibitor YVAD (25 μM) to inhibit the inflammasome for 1 h pre-infection.
C. rodentium was added at an MOI of 10:1 for 2 h; then ATP (2.5 mM) was added for 30 min. Cells were lysed using RIPA Buffer containing protease inhibitor cocktail (Sigma), then centrifuged at 18,800 g for 10 min to pellet cell debris. Samples were run on 4–15% Mini-PROTEAN
® TGX™ precast protein gels (Bio-Rad). When preparing the samples to run via electrophoresis, 2 µL of running buffer composed of 10% DTT was loaded into Eppendorf tubes each containing 10 µL of the desired cell lysate. The samples were then heated for 5 min to 50 °C and then spun at 14,000 RPM for 30 s to ensure the buffer/cell solutions were thoroughly mixed. Approximately 10 µL of solution was added to each lane of the gel, using PageRuler Prestained Ladder (ThermoFisher) as the molecular-weight marker. Electrical current was applied for 60 min at 150 V. Western blot transfer was conducted via a submersion method at 100 V and using a nitrocellulose membrane. The membrane was then blocked in 5% BSA in TBS-0.1%Tween20 for 1 h at room temperature with low agitation. Primary antibodies (Occludin, Abcam ab167161, Claudin 1, Abcam ab180158, ZO-1, Invitrogen 33-9100, and Actin, Abcam ab8226) of 1:1000 dilution were made to volumes of 5 mL using a 0.05% BSA in a TBS-0.1%Tween20 solution and the membrane was cut accordingly and probed overnight at 4 °C with low agitation. The membrane sections were then rinsed with 3 × 5 min washes using 5 mL of TBS-0.1%Tween20. Subsequently, the sections were then probed with 5 mL 1:5000 dilutions of secondary antibody (Jackson ImmunoResearch 111-035-003) in TBS-0.1%Tween20 for 45 min at room temperature with low agitation. For imaging purposes, the membrane sections were saturated with 400 µL of a 1:1 ratio of Amersham Western Blotting Detection Reagent (GE Healthcare Life Sciences, Sigma-Aldritch, Oakville, Canada). The membrane sections were then reassembled into the complete blot and imaged using 30 s exposure. Images were inverted and sharpened using FIJI [
17].
2.7. Statistical Analysis
Statistical analysis was performed using Prism9 for Mac (Graph Pad, San Diego, CA, USA). Kruskal–Wallis ANOVA with Dunn’s Multiple comparison test were used for the bar graphs. Two-way ANOVA using multiple comparisons was used to examine barrier recovery. Graphs represent the mean ±SEM and all comparisons with p < 0.05 were considered significant.
4. Discussion
The intestinal epithelial barrier is known to be an important part of the innate immune response [
22]. Not only does it provide a physical barrier to prevent infection, but epithelial cells also secrete many factors that influence both the microbiota and the immune system, such as cytokines, chemokines, and AMP. Inflammasome activation have been studied extensively in immune cells, such as macrophages and neutrophils, where the inflammasome contributes specifically to microbial clearance [
9,
11,
23]; however, the role of the NLRP3 inflammasome has not been fully characterized in epithelial cells. Evidence suggests that the inflammasome can have both detrimental or protective roles in gastrointestinal pathophysiology, depending on the immune response; detrimental in IL-10 deficiency but protective against intestinal barrier disruption [
24]. Individuals with IBD not only have a disrupted epithelial barrier, but also a dysregulated immune response that leads to a chronic inflammatory state, through poorly defined mechanisms. Some of these individuals demonstrate hypoproduction of IL-1β when exposed to potential pathogens; this dysregulation of the immune system and inability to control potential pathogens may contribute to a chronic inflammatory state [
25]. Building on our previous findings that NLRP3 can enhance bacterial killing by macrophages and, thus, reduce pro-inflammatory responses, and recognizing the complexity of the gut mucosa, the aim for this study was to assess the impact of the NLRP3 inflammasome on the complex relationship between bacteria, macrophages, and epithelial cells. In addition, we looked at the role of the NLRP3 inflammasome in epithelial cells and whether it affected response to
C. rodentium infection.
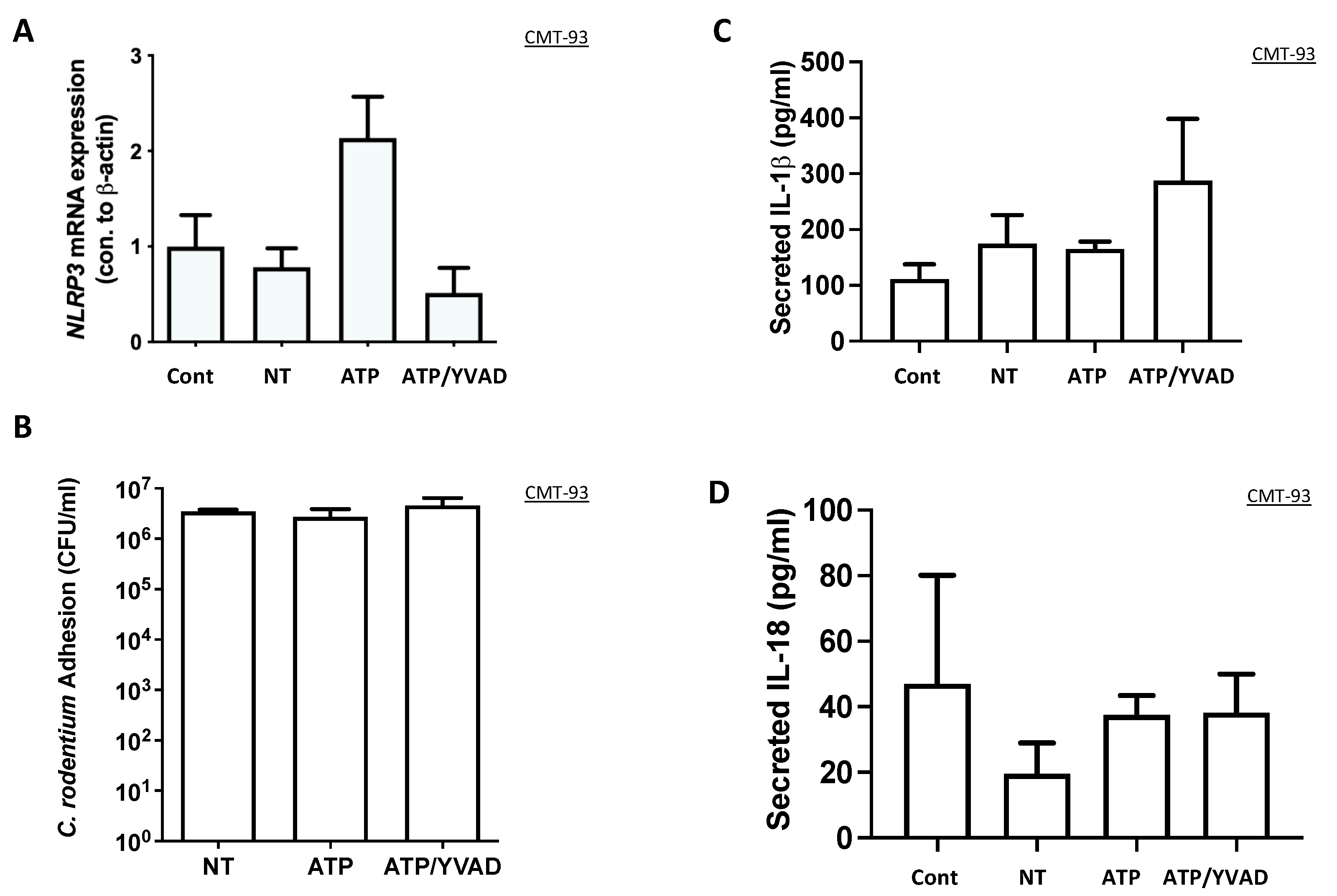
Our initial hypothesis was that epithelial cells can contribute to C. rodentium clearance directly through inflammasome activation, as shown in macrophages. Interestingly, the number of bacteria adhered to the epithelium after inflammasome activation was no different, suggesting that the NLRP3 inflammasome may not directly affect epithelial response to infection. NLRP3 gene expression was increased in ATP-activated CMT-93 cells; however, there was no significant change in IL-1β or IL-18 secretion. This suggests that ATP can induce transcription of NLRP3, but there is no activation, in contrast to macrophages, where ATP acts as the second signal for inflammasome activation. A limitation of this study is that we did not examine NLRP3 protein expression in the epithelial cell line and instead focused on the secretion of IL-18 and IL-1β as our indication of inflammasome activation; however, cytokine secretion best represents inflammasome activation.
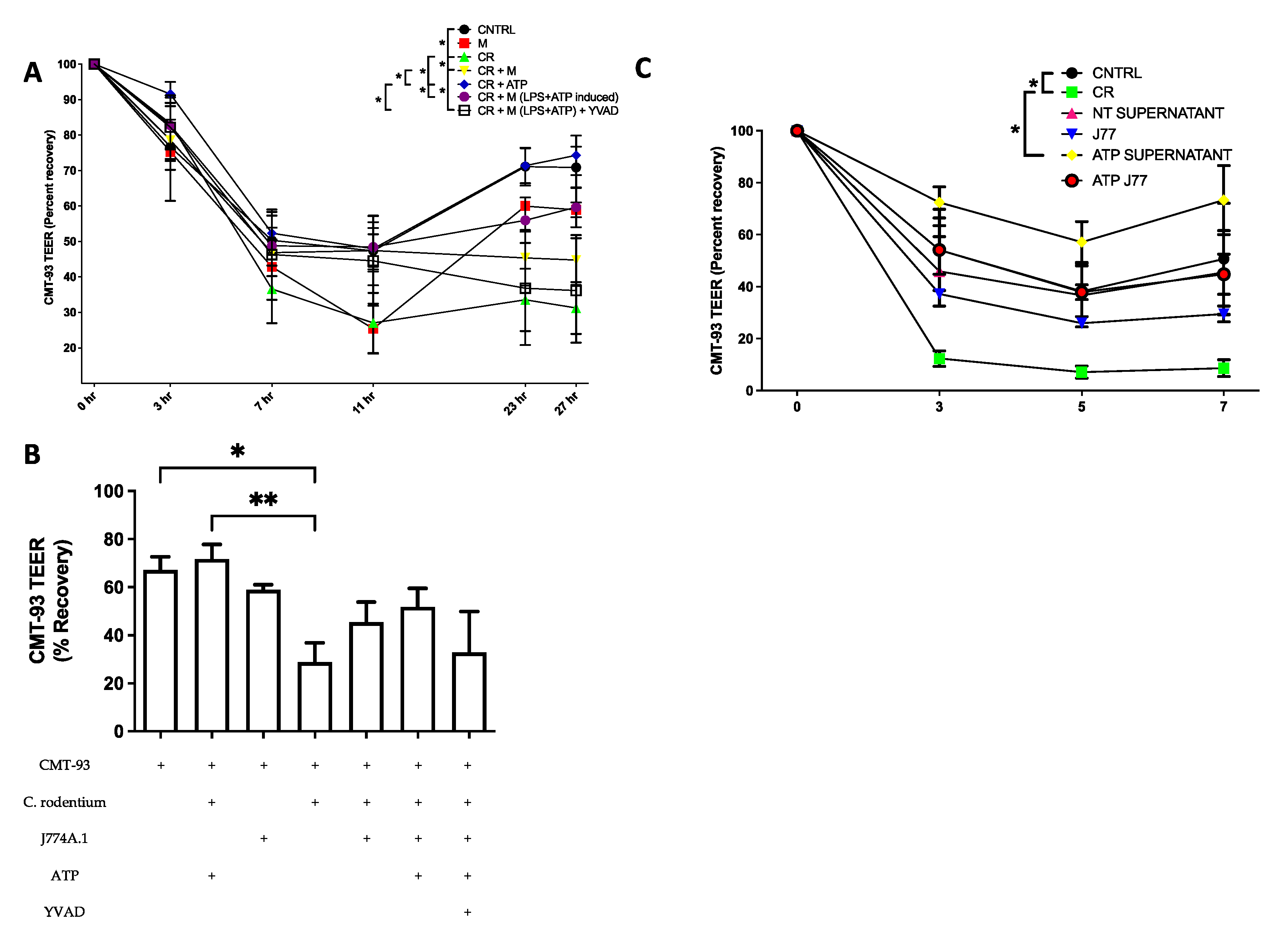
Next, we looked at the role of the inflammasome in epithelial barrier recovery from C. rodentium infection in the presence of macrophages using a multi-cellular Transwell model. We observed that the addition of ATP to the basolateral side significantly improved barrier recovery, independent of macrophages, suggesting that basolateral purinergic receptor activation induces tight junction formation. In addition, epithelial cells exposed to ATP had less adherent C. rodentium after 24 h, suggesting that activation of the epithelial purinergic receptor may have an additional role to control infection, other than activation of the inflammasome. Although not statistically significant, the presence of macrophages also improved the epithelial barrier, with ATP-induced cells showing the highest recovery with inhibition achieved using YVAD. This supports our hypothesis that NLRP3 inflammasome activation in macrophages can function in a protective manner on the epithelial barrier during enteric infections, or when managing mucosa-associated bacteria in general.
To assess if the recovery was due to the presence of the macrophages or a secreted factor, supernatants were collected from previously infected macrophages, filtered to remove bacteria, and added to the basolateral side. Marked improvements in CMT-93 epithelial barrier integrity were observed when treated with macrophage supernatants compared to macrophage cells, or when treated with ATP-stimulated macrophage supernatants, compared to ATP-stimulated macrophage cells. This suggests that a factor secreted from macrophage cells is responsible for this barrier protection seen in epithelial cells and supports future investigations to identify this factor or factors involved in this complex model.
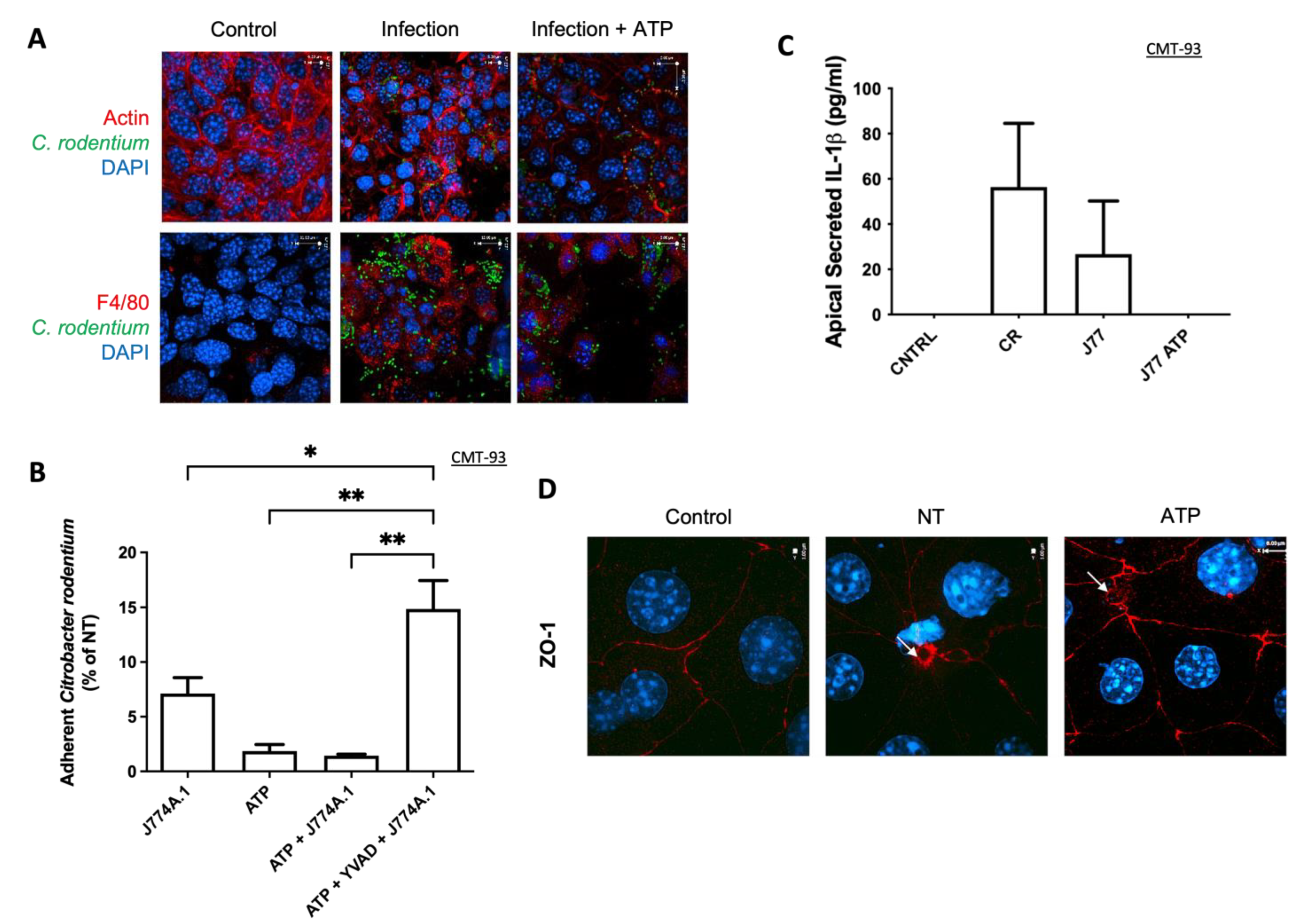
It has previously been shown that extracellular ATP acts as a chemoattractant for macrophage recruitment to the apical membrane of epithelial cells [
26]. We did not assess ATP secretion from epithelial cells, but during
C. rodentium infection, macrophages were indeed recruited to the apical membrane, whereas ATP-induced inflammasome activation had no additional effect. This suggests that the increased recovery was not due to macrophage recruitment alone, but rather by factors secreted from macrophage cells. In addition, there were less bacteria adhered to CMT-93 epithelial cells in the presence of ATP as well as inflammasome-activated macrophages. This finding not only supports our previous study, where we showed an increase in bacterial killing by ATP-activated macrophages, but also suggests that extracellular ATP can have a protective role in epithelial cells [
11]. Reduced adherence in the Transwell model contrasts with
Figure 1, where there was no effect suggesting a basolateral specific effect for ATP activation. This is likely because epithelial cells are not polarized in a standard culture dish as they are in a Transwell model.
Another defense mechanism against infection within the bowel is the epithelial barrier, which is known to be damaged or diminished in IBD patients [
27]. This barrier provides a boundary between apical and basolateral membranes as well as preventing any luminal bacteria from getting into the mucosa. Tight junction proteins anchor intestinal epithelial cells together and control paracellular trafficking by providing a semipermeable barrier. It has been previously demonstrated that macrophages can express tight junction proteins (claudin-1, ZO-1, and occludin) in an airway model, but this has not been described in the gut or gut-derived cells [
21]. Examining these tight junction proteins by western blot in J77 macrophage cells demonstrated no change in protein levels in response to ATP stimulation. We found changes in RNA without any difference in protein levels, suggesting post translational regulation either due to infection, inflammasome activation, or a combination of both. Claudins are an integral part of the intestinal epithelial barrier, where they have been shown to be involved in tethering epithelial cells together [
28]. Poor epithelial barrier integrity, or leaky gut, is considered a hallmark in IBD and a possible link to microbial-induced inflammation of the submucosa [
4,
5]. Claudin-1 has been shown to be dysregulated in Crohn disease, correlating to a leaky barrier [
29,
30]. Claudin-1 is specifically expressed in M2 macrophages, as they are immune regulatory and help resolution of inflammation. This expression has been shown to be induced by epithelial cell-secreted transforming growth factor beta (TGF-β) [
31,
32]. Our previous work illustrated that ATP-induced NLRP3 inflammasome activation leads to the macrophage cell population shift towards an M2 class of macrophage during infection with
C. rodentium [
11]. This differentiation to the M2 subtype of macrophage may, therefore, cause these cells to interact with epithelia in a manner that promotes improved barrier integrity.
Finally, we assessed whether ATP-induced NLRP3 inflammasome activation in epithelial cells would affect the ability of co-cultured macrophages to clear C. rodentium. Interestingly, treatment of CMT-93 cells with the inflammasome activator ATP did not change the ability of co-cultured macrophage cells to clear infection. This suggests that epithelial cells secrete factors (yet unidentified) independent of infection that increase macrophage clearance of a pathogen. We did not determine what this factor was but speculate that it could be a multitude of molecules, such as AMPs. This mechanism will require a more intensive study for further characterization of the signaling pathways involved.
In summary, we observed that ATP activation of the inflammasome in macrophages can have a positive impact on epithelial barrier recovery after infection with C. rodentium. These activated macrophages are recruited to the apical membrane, possibly through expression of tight junction proteins, which may promote clearance of infection. The key players in inflammasome activation in epithelial cells still need further investigation; however, we did find that extracellular ATP at the basolateral aspect can elicit epithelial barrier recovery, greater than the addition of activated macrophages. In addition, epithelial cells secrete factors constitutively that can enhance macrophage ability to clear infection. Together, this work supports inflammasome-mediated crosstalk between epithelial cells and macrophages, which promotes elimination of pathogens in the gut mucosa. Immune modulation targeting the inflammasome (perhaps enhancing its activity in this case) may be of benefit to patients with IBD, as this may lead to more efficient clearing of invading bacteria in addition to a recovery of the epithelial barrier, which could then counterbalance the hyperreactive immune response.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}