
The Role of Macrophages in Cardiac Function and Disease


Abstract
:1. Introduction
2. Crosstalk between Inflammation and Cardiac Damage: Cellular and Molecular Mechanisms
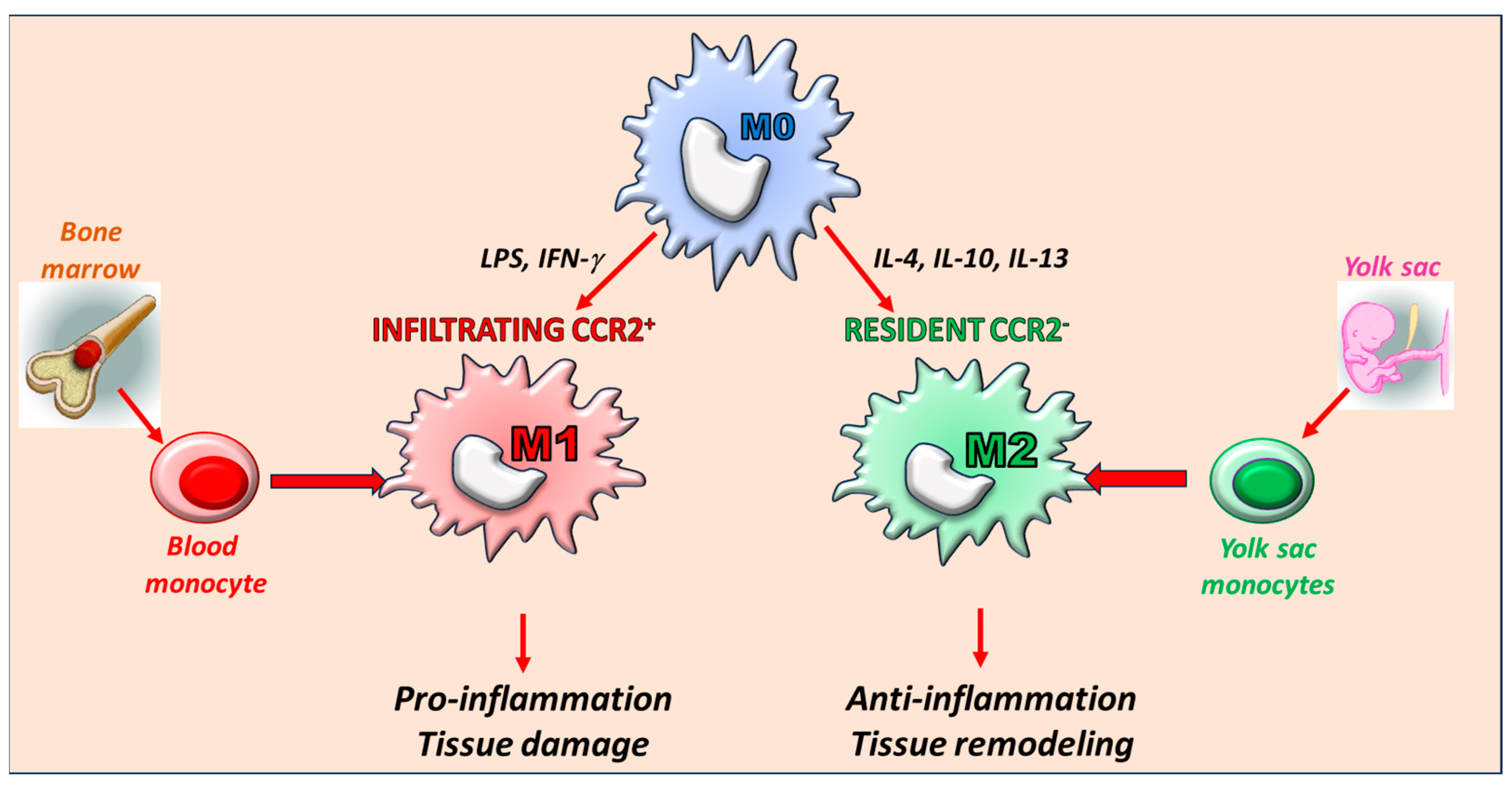
3. The Key Role of Macrophages: Origin and Phenotype
4. The Role of Macrophages in Heart Physiology
5. The Role of Macrophages in Heart Disease
5.1. Macrophages in Heart Failure
5.2. Macrophages in Anthracycline-Dependent Cardiotoxicity
5.3. Macrophages in Atherosclerosis
6. Crosstalk between Inflammation and Cardiovascular Disease: The Functional Role of NFκB
NFκB as a Potential Therapeutic Target

{kind=link}
{kind=link}
{kind=link}
| Mechanisms of NFkB Targeting | References |
|---|---|
| IKK overexpression | Refs. [84,96] |
| Deletion of the p50 subunit of NFkB | Ref. [97] |
| Overexpression of IκBα | Refs. [98,99] |
| Overexpression of a phosphorylation-resistant form of IκBα | Refs. [100,101] |
| Nuclear accumulation of IκBα by RH domain of GRK5 | Refs. [34,103] |
| Inhibition of NFκB activity by GRK2 | Refs. [33,95] |
| Pharmacologic inhibitors of NFκB | Refs. [104,105,106,107] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scheen, A.J. JUPITER: Reduction by rosuvastatin of cardiovascular events and mortality in healthy subjects without hyperlipidaemia but with elevated C-reactive protein. Rev. Med. Liege 2008, 63, 749–753. [Google Scholar] [PubMed]
- Ridker, P.M.; Group, J.S. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: Rationale and design of the JUPITER trial. Circulation 2003, 108, 2292–2297. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; De Luca, N.; Trimarco, B.; Iaccarino, G. The Antioxidant Therapy: New Insights in the Treatment of Hypertension. Front. Physiol. 2018, 9, 258. [Google Scholar] [CrossRef]
- Grailer, J.J.; Haggadone, M.D.; Sarma, J.V.; Zetoune, F.S.; Ward, P.A. Induction of M2 regulatory macrophages through the beta2-adrenergic receptor with protection during endotoxemia and acute lung injury. J. Innate Immun. 2014, 6, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Hamada, M.; Hiasa, G.; Sasaki, O.; Suzuki, M.; Hara, Y.; Shigematsu, Y.; Hiwada, K. Effect of beta-blockers on circulating levels of inflammatory and anti-inflammatory cytokines in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2001, 37, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Li, X.; Chen, G.; Liang, H.; Wu, Y.; Tong, J.; Ouyang, W. Propranolol Attenuates Surgical Stress-Induced Elevation of the Regulatory T Cell Response in Patients Undergoing Radical Mastectomy. J. Immunol. 2016, 196, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Jagadeesh, G. A potential role of the renin-angiotensin-aldosterone system in epithelial-to-mesenchymal transition-induced renal abnormalities: Mechanisms and therapeutic implications. Pharmacol. Res. 2019, 146, 104314. [Google Scholar] [CrossRef]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflam. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Bolla, G.B.; Fedele, A.; Faggiano, A.; Sala, C.; Santangelo, G.; Carugo, S. Effects of Sacubitril/Valsartan on biomarkers of fibrosis and inflammation in patients with heart failure with reduced ejection fraction. BMC Cardiovasc. Disord. 2022, 22, 217. [Google Scholar] [CrossRef]
- Jia, T.; Wang, X.; Tang, Y.; Yu, W.; Li, C.; Cui, S.; Zhu, J.; Meng, W.; Wang, C.; Wang, Q. Sacubitril Ameliorates Cardiac Fibrosis Through Inhibiting TRPM7 Channel. Front. Cell Dev. Biol. 2021, 9, 760035. [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T.; Group, C.P.I. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef]
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef]
- Gambardella, J.; Franco, A.; Giudice, C.D.; Fiordelisi, A.; Cipolletta, E.; Ciccarelli, M.; Trimarco, B.; Iaccarino, G.; Sorriento, D. Dual role of GRK5 in cancer development and progression. Transl. Med. UniSa 2016, 14, 28–37. [Google Scholar]
- Tocchetti, C.G.; Ameri, P.; de Boer, R.A.; D’Alessandra, Y.; Russo, M.; Sorriento, D.; Ciccarelli, M.; Kiss, B.; Bertrand, L.; Dawson, D.; et al. Cardiac dysfunction in cancer patients: Beyond direct cardiomyocyte damage of anticancer drugs: Novel cardio-oncology insights from the joint 2019 meeting of the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc. Res. 2020, 116, 1820–1834. [Google Scholar] [CrossRef]
- Wagner, J.U.G.; Dimmeler, S. Cellular cross-talks in the diseased and aging heart. J. Mol. Cell. Cardiol. 2020, 138, 136–146. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2020, 598, 2923–2939. [Google Scholar] [CrossRef]
- Zhang, P.; Su, J.; Mende, U. Cross talk between cardiac myocytes and fibroblasts: From multiscale investigative approaches to mechanisms and functional consequences. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1385–H1396. [Google Scholar] [CrossRef]
- Jian, Y.; Zhou, X.; Shan, W.; Chen, C.; Ge, W.; Cui, J.; Yi, W.; Sun, Y. Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell Commun. Signal 2023, 21, 109. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Williams, J.W.; Giannarelli, C.; Rahman, A.; Randolph, G.J.; Kovacic, J.C. Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 2018, 72, 2166–2180. [Google Scholar] [CrossRef]
- Hitscherich, P.; Lee, E.J. Crosstalk Between Cardiac Cells and Macrophages Postmyocardial Infarction: Insights from In Vitro Studies. Tissue Eng. Part. B Rev. 2021, 27, 475–485. [Google Scholar] [CrossRef]
- Tattersall, I.W.; Du, J.; Cong, Z.; Cho, B.S.; Klein, A.M.; Dieck, C.L.; Chaudhri, R.A.; Cuervo, H.; Herts, J.H.; Kitajewski, J. In vitro modeling of endothelial interaction with macrophages and pericytes demonstrates Notch signaling function in the vascular microenvironment. Angiogenesis 2016, 19, 201–215. [Google Scholar] [CrossRef]
- Butoi, E.; Gan, A.M.; Tucureanu, M.M.; Stan, D.; Macarie, R.D.; Constantinescu, C.; Calin, M.; Simionescu, M.; Manduteanu, I. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim. Biophys. Acta 2016, 1863, 1568–1578. [Google Scholar] [CrossRef]
- Fernando, M.R.; Giembycz, M.A.; McKay, D.M. Bidirectional crosstalk via IL-6, PGE2 and PGD2 between murine myofibroblasts and alternatively activated macrophages enhances anti-inflammatory phenotype in both cells. Br. J. Pharmacol. 2016, 173, 899–912. [Google Scholar] [CrossRef]
- Mitchell, M.D.; Laird, R.E.; Brown, R.D.; Long, C.S. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1139–H1147. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-kappaB in the heart: To be or not to NF-kappaB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Sorriento, D.; Iaccarino, G. Inflammation and Cardiovascular Diseases: The Most Recent Findings. Int. J. Mol. Sci. 2019, 20, 3879. [Google Scholar] [CrossRef]
- Sorriento, D.; Santulli, G.; Ciccarelli, M.; Maione, A.S.; Illario, M.; Trimarco, B.; Iaccarino, G. The Amino-Terminal Domain of GRK5 Inhibits Cardiac Hypertrophy through the Regulation of Calcium-Calmodulin Dependent Transcription Factors. Int. J. Mol. Sci. 2018, 19, 861. [Google Scholar] [CrossRef]
- Sorriento, D.; Santulli, G.; Franco, A.; Cipolletta, E.; Napolitano, L.; Gambardella, J.; Gomez-Monterrey, I.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J. Cardiovasc. Transl. Res. 2015, 8, 493–502. [Google Scholar] [CrossRef]
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 2010, 56, 696–704. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Cipolletta, E.; Santulli, G.; Fusco, A.; Zhou, R.H.; Eckhart, A.D.; Peppel, K.; Koch, W.J.; Trimarco, B.; et al. Impaired neoangiogenesis in beta(2)-adrenoceptor gene-deficient mice: Restoration by intravascular human beta(2)-adrenoceptor gene transfer and role of NFkappaB and CREB transcription factors. Br. J. Pharmacol. 2011, 162, 712–721. [Google Scholar] [CrossRef]
- Bolego, C.; Cignarella, A.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage function and polarization in cardiovascular disease: A role of estrogen signaling? Arter. Thromb. Vasc. Biol. 2013, 33, 1127–1134. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 macrophages and their overlaps—Myth or reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef]
- Hu, S.; Yang, M.; Huang, S.; Zhong, S.; Zhang, Q.; Ding, H.; Xiong, X.; Hu, Z.; Yang, Y. Different Roles of Resident and Non-resident Macrophages in Cardiac Fibrosis. Front. Cardiovasc. Med. 2022, 9, 818188. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Moskalik, A.; Niderla-Bielinska, J.; Ratajska, A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 2022, 27, 1413–1430. [Google Scholar] [CrossRef]
- Alvarez-Argote, S.; O’Meara, C.C. The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair. Int. J. Mol. Sci. 2021, 22, 7923. [Google Scholar] [CrossRef]
- Nielsen, M.C.; Andersen, M.N.; Moller, H.J. Monocyte isolation techniques significantly impact the phenotype of both isolated monocytes and derived macrophages in vitro. Immunology 2020, 159, 63–74. [Google Scholar] [CrossRef]
- Zaman, R.; Hamidzada, H.; Epelman, S. Exploring cardiac macrophage heterogeneity in the healthy and diseased myocardium. Curr. Opin. Immunol. 2021, 68, 54–63. [Google Scholar] [CrossRef]
- Gao, Y.; Qian, N.; Xu, J.; Wang, Y. The Roles of Macrophages in Heart Regeneration and Repair After Injury. Front. Cardiovasc. Med. 2021, 8, 744615. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Godwin, J.W.; Debuque, R.; Salimova, E.; Rosenthal, N.A. Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen. Med. 2017, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- de Couto, G. Macrophages in cardiac repair: Environmental cues and therapeutic strategies. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e520. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y.M. Efferocytosis and Its Role in Inflammatory Disorders. Front. Cell Dev. Biol. 2022, 10, 839248. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Wang, Y.; Subramanian, M.; Yurdagul, A., Jr.; Barbosa-Lorenzi, V.C.; Cai, B.; de Juan-Sanz, J.; Ryan, T.A.; Nomura, M.; Maxfield, F.R.; Tabas, I. Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 2017, 171, 331–345.e322. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Nassef, M.Z.; Hanke, J.E.; Hiller, K. Mitochondrial metabolism in macrophages. Am. J. Physiol. Cell Physiol. 2021, 321, C1070–C1081. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, D.D.; Liu, X.; Tian, R. Metabolic Modulation of Macrophage Function Post Myocardial Infarction. Front. Physiol. 2020, 11, 674. [Google Scholar] [CrossRef]
- Chen, G.; Jiang, H.; Yao, Y.; Tao, Z.; Chen, W.; Huang, F.; Chen, X. Macrophage, a potential targeted therapeutic immune cell for cardiomyopathy. Front. Cell Dev. Biol. 2022, 10, 908790. [Google Scholar] [CrossRef]
- Duerr, G.D.; Heinemann, J.C.; Suchan, G.; Kolobara, E.; Wenzel, D.; Geisen, C.; Matthey, M.; Passe-Tietjen, K.; Mahmud, W.; Ghanem, A.; et al. The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic. Res. Cardiol. 2014, 109, 425. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Yang, Q.; et al. Self-Maintenance of Cardiac Resident Reparative Macrophages Attenuates Doxorubicin-Induced Cardiomyopathy Through the SR-A1-c-Myc Axis. Circ. Res. 2020, 127, 610–627. [Google Scholar] [CrossRef]
- Prevete, N.; Poto, R.; Marone, G.; Varricchi, G. Unleashing the power of formyl peptide receptor 2 in cardiovascular disease. Cytokine 2023, 169, 156298. [Google Scholar] [CrossRef]
- Wang, M.; Qian, L.; Li, J.; Ming, H.; Fang, L.; Li, Y.; Zhang, M.; Xu, Y.; Ban, Y.; Zhang, W.; et al. GHSR deficiency exacerbates cardiac fibrosis: Role in macrophage inflammasome activation and myofibroblast differentiation. Cardiovasc. Res. 2020, 116, 2091–2102. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure. JACC Basic. Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Lim, G.B. Heart failure: Macrophages promote cardiac fibrosis and diastolic dysfunction. Nat. Rev. Cardiol. 2018, 15, 196–197. [Google Scholar] [CrossRef]
- Xia, Y.; Lee, K.; Li, N.; Corbett, D.; Mendoza, L.; Frangogiannis, N.G. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem. Cell Biol. 2009, 131, 471–481. [Google Scholar] [CrossRef]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic. Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. (Warsz) 2009, 57, 165–176. [Google Scholar] [CrossRef]
- Mai, W.; Liao, Y. Targeting IL-1beta in the Treatment of Atherosclerosis. Front. Immunol. 2020, 11, 589654. [Google Scholar] [CrossRef]
- Sager, H.B.; Heidt, T.; Hulsmans, M.; Dutta, P.; Courties, G.; Sebas, M.; Wojtkiewicz, G.R.; Tricot, B.; Iwamoto, Y.; Sun, Y.; et al. Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 2015, 132, 1880–1890. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Nozaki, N.; Shishido, T.; Takeishi, Y.; Kubota, I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation 2004, 110, 2869–2874. [Google Scholar] [CrossRef]
- Pecoraro, M.; Del Pizzo, M.; Marzocco, S.; Sorrentino, R.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2016, 293, 44–52. [Google Scholar] [CrossRef]
- Shaker, R.A.; Abboud, S.H.; Assad, H.C.; Hadi, N. Enoxaparin attenuates doxorubicin induced cardiotoxicity in rats via interfering with oxidative stress, inflammation and apoptosis. BMC Pharmacol. Toxicol. 2018, 19, 3. [Google Scholar] [CrossRef]
- Gambardella, J.; Fiordelisi, A.; Cerasuolo, F.A.; Buonaiuto, A.; Avvisato, R.; Viti, A.; Sommella, E.; Merciai, F.; Salviati, E.; Campiglia, P.; et al. Experimental evidence and clinical implications of Warburg effect in the skeletal muscle of Fabry disease. iScience 2023, 26, 106074. [Google Scholar] [CrossRef]
- Gan, J.; Guo, L.; Zhang, X.; Yu, Q.; Yang, Q.; Zhang, Y.; Zeng, W.; Jiang, X.; Guo, M. Anti-inflammatory therapy of atherosclerosis: Focusing on IKKbeta. J. Inflamm. 2023, 20, 8. [Google Scholar] [CrossRef]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. [Google Scholar] [CrossRef]
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Matsumori, A. Nuclear Factor-kappaB is a Prime Candidate for the Diagnosis and Control of Inflammatory Cardiovascular Disease. Eur. Cardiol. 2023, 18, e40. [Google Scholar] [CrossRef]
- Mussbacher, M.; Derler, M.; Basilio, J.; Schmid, J.A. NF-kappaB in monocytes and macrophages—An inflammatory master regulator in multitalented immune cells. Front. Immunol. 2023, 14, 1134661. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.; Page, S.; Rogler, G.; Bartsch, A.; Brandl, R.; Knuechel, R.; Page, M.; Kaltschmidt, C.; Baeuerle, P.A.; Neumeier, D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Investig. 1996, 97, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Forster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 2003, 112, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Heo, Y.J.; Choi, S.E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Anti-inflammatory Effects of Empagliflozin and Gemigliptin on LPS-Stimulated Macrophage via the IKK/NF-kappaB, MKK7/JNK, and JAK2/STAT1 Signalling Pathways. J. Immunol. Res. 2021, 2021, 9944880. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schonbeck, U.; Libby, P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arter. Thromb. Vasc. Biol. 2006, 26, 611–617. [Google Scholar] [CrossRef]
- Maier, H.J.; Schips, T.G.; Wietelmann, A.; Kruger, M.; Brunner, C.; Sauter, M.; Klingel, K.; Bottger, T.; Braun, T.; Wirth, T. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 11794–11799. [Google Scholar] [CrossRef]
- Kawano, S.; Kubota, T.; Monden, Y.; Tsutsumi, T.; Inoue, T.; Kawamura, N.; Tsutsui, H.; Sunagawa, K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1337–H1344. [Google Scholar] [CrossRef] [PubMed]
- Zelarayan, L.; Renger, A.; Noack, C.; Zafiriou, M.P.; Gehrke, C.; van der Nagel, R.; Dietz, R.; de Windt, L.; Bergmann, M.W. NF-kappaB activation is required for adaptive cardiac hypertrophy. Cardiovasc. Res. 2009, 84, 416–424. [Google Scholar] [CrossRef]
- Trescher, K.; Bernecker, O.; Fellner, B.; Gyongyosi, M.; Krieger, S.; Demartin, R.; Wolner, E.; Podesser, B.K. Adenovirus-mediated overexpression of inhibitor kappa B-alpha attenuates postinfarct remodeling in the rat heart. Eur. J. Cardiothorac. Surg. 2004, 26, 960–967. [Google Scholar] [CrossRef]
- Hamid, T.; Guo, S.Z.; Kingery, J.R.; Xiang, X.; Dawn, B.; Prabhu, S.D. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc. Res. 2011, 89, 129–138. [Google Scholar] [CrossRef]
- Han, M.; Chen, X.C.; Sun, M.H.; Gai, M.T.; Yang, Y.N.; Gao, X.M.; Ma, X.; Chen, B.D.; Ma, Y.T. Overexpression of IkappaBalpha in cardiomyocytes alleviates hydrogen peroxide-induced apoptosis and autophagy by inhibiting NF-kappaB activation. Lipids Health Dis. 2020, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; De Rosa, M.; Sala, M.; Pacelli, R.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. A Novel Small Peptide Inhibitor of NFkappaB, RH10, Blocks Oxidative Stress-Dependent Phenotypes in Cancer. Oxid. Med. Cell. Longev. 2018, 2018, 5801807. [Google Scholar] [CrossRef] [PubMed]
- Karunaweera, N.; Raju, R.; Gyengesi, E.; Munch, G. Plant polyphenols as inhibitors of NF-kappaB induced cytokine production-a potential anti-inflammatory treatment for Alzheimer’s disease? Front. Mol. Neurosci. 2015, 8, 24. [Google Scholar] [CrossRef]
- Sorriento, D.; Illario, M.; Finelli, R.; Iaccarino, G. To NFkappaB or not to NFkappaB: The Dilemma on How to Inhibit a Cancer Cell Fate Regulator. Transl. Med. UniSa 2012, 4, 73–85. [Google Scholar]
- Zanotto-Filho, A.; Delgado-Canedo, A.; Schroder, R.; Becker, M.; Klamt, F.; Moreira, J.C. The pharmacological NFkappaB inhibitors BAY117082 and MG132 induce cell arrest and apoptosis in leukemia cells through ROS-mitochondria pathway activation. Cancer Lett. 2010, 288, 192–203. [Google Scholar] [CrossRef]



| Cardiovascular Pathologies Affected by NFkB |
|---|
| Myocarditis |
| Myocardial infarction |
| Left ventricular hypertrophy |
| Cardiac inflammation |
| Diabetic cardiomyopathy |
| Atherosclerosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prevete, N.; Sorriento, D. The Role of Macrophages in Cardiac Function and Disease. J. Mol. Pathol. 2023, 4, 318-332. https://doi.org/10.3390/jmp4040026
Prevete N, Sorriento D. The Role of Macrophages in Cardiac Function and Disease. Journal of Molecular Pathology. 2023; 4(4):318-332. https://doi.org/10.3390/jmp4040026
Chicago/Turabian StylePrevete, Nella, and Daniela Sorriento. 2023. "The Role of Macrophages in Cardiac Function and Disease" Journal of Molecular Pathology 4, no. 4: 318-332. https://doi.org/10.3390/jmp4040026