
Liquid Biopsy in Advanced Colorectal Cancer: Clinical Applications of Different Analytes
 , , ,
, , ,
Abstract
:1. Introduction
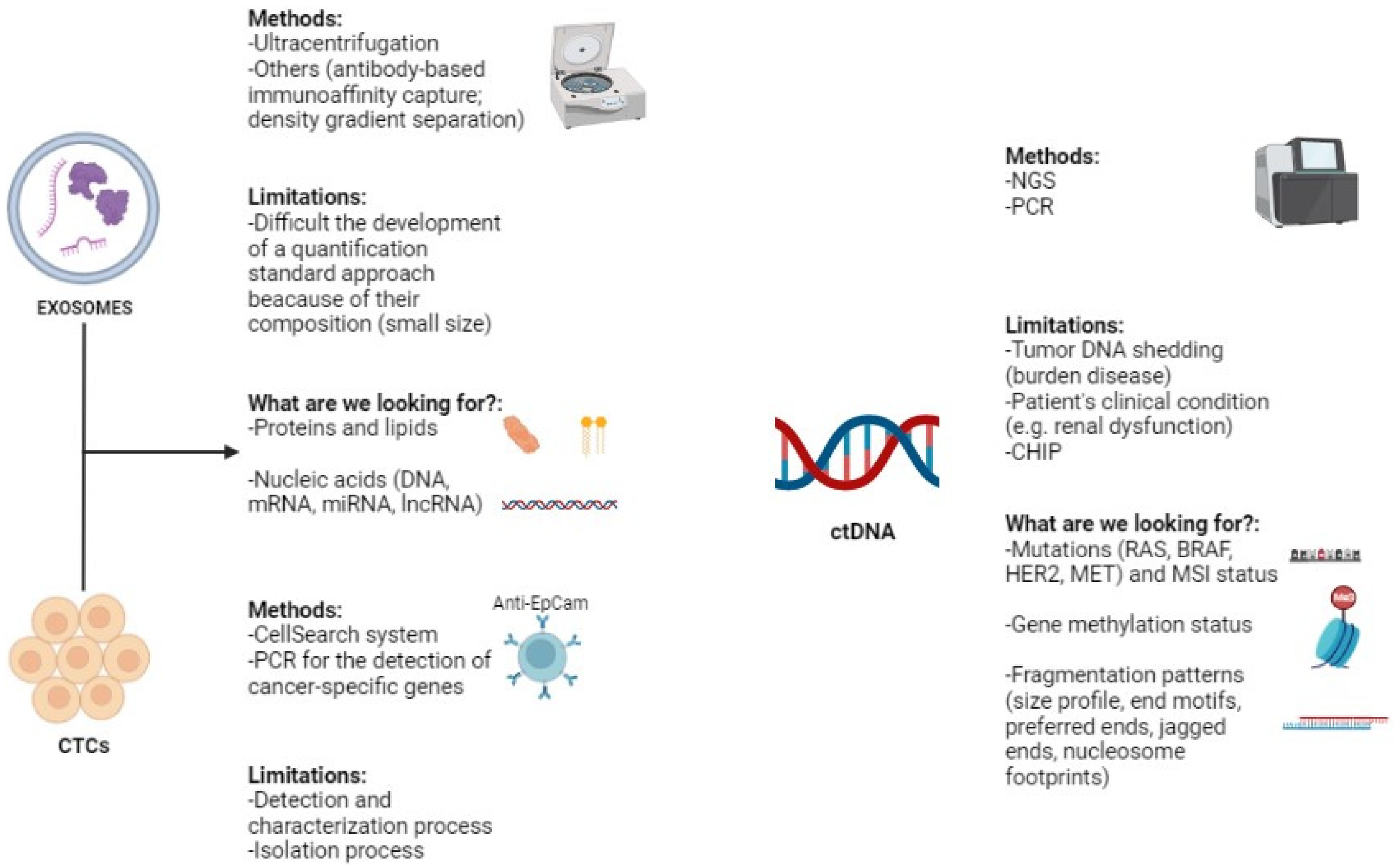
2. Exosomes
2.1. Introduction, Methodology, and Limitations
2.2. Diagnosis, Prognosis, and Therapeutic Response Evaluation
3. Circulating Tumor Cells
3.1. Introduction, Methodology, and Limitations
3.2. Metastatic Diagnosis, Prognosis, and Therapeutic Response Evaluation
4. Circulating Tumor DNA
4.1. Introduction, Methodology, and Limitations
4.2. Metastatic Diagnosis and Prognosis
4.3. Therapeutic Response Evaluation
4.4. Detection of RAS Mutations
4.5. Rechallenge with Anti-EGR Antibodies
4.6. Detection of Other Mutations (MSI, BRAF, MET, and ERBB2)
4.6.1. MSI
4.6.2. BRAF
4.6.3. MET
4.6.4. ERBB2 (HER2)
4.7. Cell-Free DNA Fragmentomics
5. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T.; et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 10–32. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 28 January 2021).
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.W.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.; Ou, S.I.; Yang, Y. NTRK fusion positive colorectal cancer is a unique subset of CRC with high TMB and microsatellite instability. Cancer Med. 2022, 11, 2541–2549. [Google Scholar] [CrossRef]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castaños-Vélez, E.; Blumenstein, B.A.; Rösch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014, 63, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pan, B.; Sun, L.; Chen, X.; Zeng, K.; Hu, X.; Xu, T.; Xu, M.; Wang, S. Circulating Exosomal miR-27a and miR-130a Act as Novel Diagnostic and Prognostic Biomarkers of Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2018, 27, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncarelli Flores, B.C.; Souza E Silva, V.; Ali Abdallah, E.; Mello, C.A.L.; Gobo Silva, M.L.; Gomes Mendes, G.; Camila Braun, A.; Aguiar Junior, S.; Thomé Domingos Chinen, L. Molecular and Kinetic Analyses of Circulating Tumor Cells as Predictive Markers of Treatment Response in Locally Advanced Rectal Cancer Patients. Cells 2019, 8, 641. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ren, L.; Li, S.; Li, W.; Zheng, X.; Yang, Y.; Fu, W.; Yi, J.; Wang, J.; Du, G. The biology, function, and applications of exosomes in cancer. Acta Pharm. Sin. B 2021, 11, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Zhang, R.; Li, Z.; Li, J. Clinical and biological significance of circulating tumor cells, circulating tumor DNA, and exosomes as biomarkers in colorectal cancer. Oncotarget 2017, 8, 55632–55645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Janku, F.; Zhan, Q.; Fan, J.B. Accessing Genetic Information with Liquid Biopsies. Trends Genet. 2015, 31, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Halvaei, S.; Daryani, S.; Eslami-S, Z.; Samadi, T.; Jafarbeik-Iravani, N.; Bakhshayesh, T.O.; Majidzadeh-A, K.; Esmaeili, R. Exosomes in Cancer Liquid Biopsy: A Focus on Breast Cancer. Mol. Ther. Nucleic Acids 2018, 10, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Eylem, C.C.; Yilmaz, M.; Derkus, B.; Nemutlu, E.; Camci, C.B.; Yilmaz, E.; Turkoglu, M.A.; Aytac, B.; Ozyurt, N.; Emregul, E. Untargeted multi-omic analysis of colorectal cancer-specific exosomes reveals joint pathways of colorectal cancer in both clinical samples and cell culture. Cancer Lett. 2020, 469, 186–194. [Google Scholar] [CrossRef]
- Galbiati, S.; Damin, F.; Burgio, V.; Brisci, A.; Soriani, N.; Belcastro, B.; Di Resta, C.; Gianni, L.; Chiari, M.; Ronzoni, M.; et al. Evaluation of three advanced methodologies, COLD-PCR, microarray and ddPCR, for identifying the mutational status by liquid biopsies in metastatic colorectal cancer patients. Clin. Chim. Acta 2019, 489, 136–143. [Google Scholar] [CrossRef]
- Kang, J.K.; Heo, S.; Kim, H.P.; Song, S.H.; Yun, H.; Han, S.W.; Kang, G.H.; Bang, D.; Kim, T.Y. Liquid biopsy-based tumor profiling for metastatic colorectal cancer patients with ultra-deep targeted sequencing. PLoS ONE 2020, 15, e0232754. [Google Scholar] [CrossRef]
- Yu, H.; Han, L.; Yuan, J.; Sun, Y. Circulating tumor cell free DNA from plasma and urine in the clinical management of colorectal cancer. Cancer Biomark. 2020, 27, 29–37. [Google Scholar] [CrossRef]
- Baassiri, A.; Nassar, F.; Mukherji, D.; Shamseddine, A.; Nasr, R.; Temraz, S. Exosomal Non Coding RNA in LIQUID Biopsies as a Promising Biomarker for Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 1398. [Google Scholar] [CrossRef] [Green Version]
- Greening, D.W.; Gopal, S.K.; Mathias, R.A.; Liu, L.; Sheng, J.; Zhu, H.J.; Simpson, R.J. Emerging roles of exosomes during epithelial-mesenchymal transition and cancer progression. Semin. Cell Dev. Biol. 2015, 40, 60–71. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Zijlstra, A.; Rubin, M.A.; Di Vizio, D. Extracellular vesicles for liquid biopsy in prostate cancer: Where are we and where are we headed? Prostate Cancer Prostatic Dis. 2017, 20, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.; de Vrij, J.; van der Vlist, E.J.; Geragousian, B.; van Bloois, L.; Mastrobattista, E.; Schiffelers, R.M.; Wauben, M.H.; Broekman, M.L.; Nolte-’t Hoen, E.N. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. J. Control. Release 2015, 200, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganig, N.; Baenke, F.; Thepkaysone, M.L.; Lin, K.; Rao, V.S.; Wong, F.C.; Polster, H.; Schneider, M.; Helm, D.; Pecqueux, M.; et al. Proteomic Analyses of Fibroblast- and Serum-Derived Exosomes Identify QSOX1 as a Marker for Non-invasive Detection of Colorectal Cancer. Cancers 2021, 13, 1351. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Guo, X.; Zhou, L.; Jia, Z.; Peng, Z.; Tang, Y.; Liu, W.; Zhu, B.; Wang, L.; et al. GPC1 exosome and its regulatory miRNAs are specific markers for the detection and target therapy of colorectal cancer. J. Cell. Mol. Med. 2017, 21, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, Y.; Zhou, Y.; Ng, T.K.; Zhao, C.; Gan, Q.; Gu, X.; Xiang, J. Circulating exosomal CPNE3 as a diagnostic and prognostic biomarker for colorectal cancer. J. Cell. Physiol. 2019, 234, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.S.; Chang, W.M.; Chen, Y.Y.; Lin, Y.F.; Liao, H.F.; Chen, C.Y. Circulating microRNA-762 upregulation in colorectal cancer may be accompanied by Wnt-1/β-catenin signaling. Cancer Biomark. 2021, 32, 111–122. [Google Scholar] [CrossRef]
- Radwan, E.; Shaltout, A.S.; Mansor, S.G.; Shafik, E.A.; Abbas, W.A.; Shehata, M.R.; Ali, M. Evaluation of circulating microRNAs-211 and 25 as diagnostic biomarkers of colorectal cancer. Mol. Biol. Rep. 2021, 48, 4601–4610. [Google Scholar] [CrossRef]
- Silva, C.M.S.; Barros-Filho, M.C.; Wong, D.V.T.; Mello, J.B.H.; Nobre, L.M.S.; Wanderley, C.W.S.; Lucetti, L.T.; Muniz, H.A.; Paiva, I.K.D.; Kuasne, H.; et al. Circulating let-7e-5p, miR-106a-5p, miR-28-3p, and miR-542-5p as a Promising microRNA Signature for the Detection of Colorectal Cancer. Cancers 2021, 13, 1493. [Google Scholar] [CrossRef]
- Karimi, N.; Ali Hosseinpour Feizi, M.; Safaralizadeh, R.; Hashemzadeh, S.; Baradaran, B.; Shokouhi, B.; Teimourian, S. Serum overexpression of miR-301a and miR-23a in patients with colorectal cancer. J. Chin. Med. Assoc. 2019, 82, 215–220. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Iinuma, H.; Yagi, T.; Matsuda, K.; Hashiguchi, Y. Circulating exosomal MicroRNA-21 as a biomarker in each tumor stage of colorectal cancer. Oncology 2017, 92, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, X.; Pan, B.; Hu, X.; Zhu, Y.; Su, Y.; Guo, Z.; Zhang, G.; Xu, M.; Xu, X.; et al. Serum exosomal miR-122 as a potential diagnostic and prognostic biomarker of colorectal cancer with liver metastasis. J. Cancer 2020, 11, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhao, Y.; Song, X.; Song, X.; Niu, L.; Xie, L. Tumor-derived exosomal miRNA-320d as a biomarker for metastatic colorectal cancer. J. Clin. Lab. Anal. 2019, 33, e23004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchetti, D.; Zurlo, I.V.; Colella, F.; Ricciardi-Tenore, C.; Di Salvatore, M.; Tortora, G.; De Maria, R.; Giuliante, F.; Cassano, A.; Basso, M.; et al. Mutational status of plasma exosomal KRAS predicts outcome in patients with metastatic colorectal cancer. Sci. Rep. 2021, 11, 22686. [Google Scholar] [CrossRef]
- Raza, A.; Khan, A.Q.; Inchakalody, V.P.; Mestiri, S.; Yoosuf, Z.S.K.M.; Bedhiafi, T.; El-Ella, D.M.A.; Taib, N.; Hydrose, S.; Akbar, S.; et al. Dynamic liquid biopsy components as predictive and prognostic biomarkers in colorectal cancer. J. Exp. Clin. Cancer Res. 2022, 41, 99. [Google Scholar] [CrossRef]
- Barbagallo, C.; Brex, D.; Caponnetto, A.; Cirnigliaro, M.; Scalia, M.; Magnano, A.; Caltabiano, R.; Barbagallo, D.; Biondi, A.; Cappellani, A.; et al. LncRNA UCA1, Upregulated in CRC Biopsies and Downregulated in Serum Exosomes, Controls mRNA Expression by RNA-RNA Interactions. Mol. Ther. Nucleic Acids 2018, 12, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, X.; Gao, S.; Jing, F.; Yang, Y.; Du, L.; Zheng, G.; Li, P.; Li, C.; Wang, C. Exosomal long noncoding RNA CRNDE-h as a novel serum-based biomarker for diagnosis and prognosis of colorectal cancer. Oncotarget 2016, 7, 85551–85563. [Google Scholar] [CrossRef]
- Pan, B.; Qin, J.; Liu, X.; He, B.; Wang, X.; Pan, Y.; Sun, H.; Xu, T.; Xu, M.; Chen, X.; et al. Identification of Serum Exosomal hsa-circ-0004771 as a Novel Diagnostic Biomarker of Colorectal Cancer. Front. Genet. 2019, 10, 1096. [Google Scholar] [CrossRef] [Green Version]
- Cekaite, L.; Eide, P.W.; Lind, G.E.; Skotheim, R.I.; Lothe, R.A. MicroRNAs as growth regulators, their function and biomarker status in colorectal cancer. Oncotarget 2016, 7, 6476–6505. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Chen, Y.; Yang, J.; Zhuo, C.; Huang, S.; Zhang, H.; Shi, Y. Clinical Perspectives on Liquid Biopsy in Metastatic Colorectal Cancer. Front. Genet. 2021, 12, 634642. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, Y.; Xu, L.; Zhan, S.; Xiao, Y.; Gao, Y.; Wu, B.; Ge, W. Protein content and functional characteristics of serum-purified exosomes from patients with colorectal cancer revealed by quantitative proteomics. Int. J. Cancer 2017, 140, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liu, J.; Li, X.; Tian, R.; Shang, K.; Dong, X.; Cao, B. Angiogenesis is promoted by exosomal DPP4 derived from 5-fluorouracil-resistant colon cancer cells. Cancer Lett. 2021, 497, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Sugimachi, K.; Iinuma, H.; Takahashi, Y.; Kurashige, J.; Sawada, G.; Ueda, M.; Uchi, R.; Ueo, H.; Takano, Y.; et al. Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br. J. Cancer 2015, 113, 275–281. [Google Scholar] [CrossRef]
- Takano, Y.; Masuda, T.; Iinuma, H.; Yamaguchi, R.; Sato, K.; Tobo, T.; Hirata, H.; Kuroda, Y.; Nambara, S.; Hayashi, N.; et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget 2017, 8, 78598–78613. [Google Scholar] [CrossRef] [Green Version]
- Fu, F.; Jiang, W.; Zhou, L.; Chen, Z. Circulating Exosomal miR-17-5p and miR-92a-3p Predict Pathologic Stage and Grade of Colorectal Cancer. Transl. Oncol. 2018, 11, 221–232. [Google Scholar] [CrossRef]
- De Miguel Pérez, D.; Rodriguez Martínez, A.; Ortigosa Palomo, A.; Delgado Ureña, M.; Garcia Puche, J.L.; Robles Remacho, A.; Exposito Hernandez, J.; Lorente Acosta, J.A.; Ortega Sánchez, F.G.; Serrano, M.J. Extracellular vesicle-miRNAs as liquid biopsy biomarkers for disease identification and prognosis in metastatic colorectal cancer patients. Sci. Rep. 2020, 10, 3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef]
- Mannavola, F.; Salerno, T.; Passarelli, A.; Tucci, M.; Internò, V.; Silvestris, F. Revisiting the Role of Exosomes in Colorectal Cancer: Where Are We Now? Front. Oncol. 2019, 9, 521. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 2018, 100, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Yan, C.; Mu, L.; Mi, Y.L.; Zhao, H.; Hu, H.; Li, X.L.; Tao, D.D.; Wu, Y.Q.; Gong, J.P.; et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 1951–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhome, R.; Goh, R.W.; Bullock, M.D.; Pillar, N.; Thirdborough, S.M.; Mellone, M.; Mirnezami, R.; Galea, D.; Veselkov, K.; Gu, Q.; et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging 2017, 9, 2666–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Lin, B.; Zhang, X.; Peng, Y.; Ye, Z.; Ma, Y.; Liang, Y.; Cao, L.; Li, X.; Li, R.; et al. Maintenance of cancer stemness by miR-196b-5p contributes to chemoresistance of colorectal cancer cells via activating STAT3 signaling pathway. Oncotarget 2017, 8, 49807–49823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Liu, Y.; Zhang, J.; Bian, Z.; Yao, S.; Fei, B.; Zhou, L.; Yin, Y.; Huang, Z. A panel of serum exosomal microRNAs as predictive markers for chemoresistance in advanced colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 315–325. [Google Scholar] [CrossRef]
- Yang, Y.N.; Zhang, R.; Du, J.W.; Yuan, H.H.; Li, Y.J.; Wei, X.L.; Du, X.X.; Jiang, S.L.; Han, Y. Predictive role of UCA1-containing exosomes in cetuximab-resistant colorectal cancer. Cancer Cell Int. 2018, 18, 164. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnol. 2020, 18, 10. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Gong, C.; Wang, Z.; Xia, Q.; Gu, F.; Hu, C.; Zhang, L.; Guo, H.; Gao, S. A33 antibody-functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomedicine 2018, 14, 1973–1985. [Google Scholar] [CrossRef]
- Jiang, M.; Jin, S.; Han, J.; Li, T.; Shi, J.; Zhong, Q.; Li, W.; Tang, W.; Huang, Q.; Zong, H. Detection and clinical significance of circulating tumor cells in colorectal cancer. Biomark. Res. 2021, 9, 85. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- De Renzi, G.; De Marco, G.; De Meo, M.; Del Rosso, E.; Gazzaniga, P.; Nicolazzo, C. In vitro cultures of circulating tumor cells: A potential tool to unravel drug sensitivity. Cancer Drug Resist. 2022, 5, 245–260. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Garrido-Navas, C.; de Miguel-Perez, D.; Exposito-Hernandez, J.; Bayarri, C.; Amezcua, V.; Ortigosa, A.; Valdivia, J.; Guerrero, R.; Garcia Puche, J.L.; Lorente, J.A.; et al. Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells 2019, 8, 1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Patelli, G.; Vaghi, C.; Tosi, F.; Mauri, G.; Amatu, A.; Massihnia, D.; Ghezzi, S.; Bonazzina, E.; Bencardino, K.; Cerea, G.; et al. Liquid Biopsy for Prognosis and Treatment in Metastatic Colorectal Cancer: Circulating Tumor Cells vs Circulating Tumor DNA. Target Oncol. 2021, 16, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; Fritsche, H.; Müller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Jänicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Sastre, J.; Maestro, M.L.; Puente, J.; Veganzones, S.; Alfonso, R.; Rafael, S.; García-Saenz, J.A.; Vidaurreta, M.; Martín, M.; Arroyo, M.; et al. Circulating tumor cells in colorectal cancer: Correlation with clinical and pathological variables. Ann. Oncol. 2008, 19, 935–938. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Tsai, W.S.; You, J.F.; Hung, H.Y.; Hsieh, P.S.; Hsieh, B.; Lenz, H.J.; Idos, G.; Friedland, S.; Yi-Jiun Pan, J.; Shao, H.J.; et al. Novel Circulating Tumor Cell Assay for Detection of Colorectal Adenomas and Cancer. Clin. Transl. Gastroenterol. 2019, 10, e00088. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Camera, S.; Akin Telli, T.; Woff, E.; Vandeputte, C.; Kehagias, P.; Guiot, T.; Critchi, G.; Wissam, Y.; Bregni, G.; Trevisi, E.; et al. Prognostic Value of the Pace of Tumor Progression as Assessed by Serial 18F-FDG PET/CT Scan and Liquid Biopsy in Refractory Colorectal Cancer: The CORIOLAN Trial. Cancers 2020, 12, 2752. [Google Scholar] [CrossRef] [PubMed]
- Arrazubi, V.; Mata, E.; Antelo, M.L.; Tarifa, A.; Herrera, J.; Zazpe, C.; Teijeira, L.; Viudez, A.; Suárez, J.; Hernández, I.; et al. Circulating Tumor Cells in Patients Undergoing Resection of Colorectal Cancer Liver Metastases. Clinical Utility for Long-Term Outcome: A Prospective Trial. Ann. Surg. Oncol. 2019, 26, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.J.; Hong, H.J.; Sun, J.; Yu, C.R.; Liu, H.S.; Li, P.Y.; Zheng, M.H. Detection and Clinical Value of Circulating Tumor Cells as an Assisted Prognostic Marker in Colorectal Cancer Patients. Cancer Manag. Res. 2021, 13, 4567–4578. [Google Scholar] [CrossRef]
- Tan, Y.; Wu, H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2018, 42, 95–106. [Google Scholar] [CrossRef]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef]
- Delgado-Ureña, M.; Ortega, F.G.; de Miguel-Pérez, D.; Rodriguez-Martínez, A.; García-Puche, J.L.; Ilyine, H.; Lorente, J.A.; Exposito-Hernandez, J.; Garrido-Navas, M.C.; Delgado-Ramirez, M.; et al. Circulating tumor cells criteria (CyCAR) versus standard RECIST criteria for treatment response assessment in metastatic colorectal cancer patients. J. Transl. Med. 2018, 16, 251. [Google Scholar] [CrossRef] [Green Version]
- Aranda, E.; Viéitez, J.M.; Gómez-España, A.; Gil Calle, S.; Salud-Salvia, A.; Graña, B.; Garcia-Alfonso, P.; Rivera, F.; Quintero-Aldana, G.A.; Reina-Zoilo, J.J.; et al. Spanish Cooperative Group for the Treatment of Digestive Tumors (TTD). FOLFOXIRI plus bevacizumab versus FOLFOX plus bevacizumab for patients with metastatic colorectal cancer and ≥3 circulating tumour cells: The randomised phase III VISNÚ-1 trial. ESMO Open 2020, 5, e000944. [Google Scholar] [CrossRef]
- Souza E Silva, V.; Chinen, L.T.; Abdallah, E.A.; Damascena, A.; Paludo, J.; Chojniak, R.; Dettino, A.L.; de Mello, C.A.; Alves, V.S.; Fanelli, M.F. Early detection of poor outcome in patients with metastatic colorectal cancer: Tumor kinetics evaluated by circulating tumor cells. Onco Targets Ther. 2016, 9, 7503–7513. [Google Scholar] [CrossRef] [Green Version]
- Cayrefourcq, L.; Thomas, F.; Mazard, T.; Assenat, E.; Assou, S.; Alix-Panabières, C. Selective treatment pressure in colon cancer drives the molecular profile of resistant circulating tumor cell clones. Mol. Cancer 2021, 20, 30. [Google Scholar] [CrossRef] [PubMed]
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802–1810. [Google Scholar] [CrossRef] [Green Version]
- Vasseur, A.; Kiavue, N.; Bidard, F.C.; Pierga, J.Y.; Cabel, L. Clinical utility of circulating tumor cells: An update. Mol. Oncol. 2021, 15, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- MANDEL, P.; METAIS, P. Les acides nucléiques du plasma sanguin chez l’homme [Nuclear Acids In Human Blood Plasma]. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating Cell-Free DNA and Colorectal Cancer: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 3356. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.I.; Nagasaka, M.; Zhu, V.W. Liquid Biopsy to Identify Actionable Genomic Alterations. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 978–997. [Google Scholar] [CrossRef]
- Bando, H.; Nakamura, Y.; Taniguchi, H.; Shiozawa, M.; Yasui, H.; Esaki, T.; Ohta, T.; Denda, T.; Satoh, T.; Yamazakiet, K.; et al. Impact of a metastatic site on circulating tumor DNA (ctDNA) analysis in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39 (Suppl. 15), 3554. [Google Scholar] [CrossRef]
- Sefrioui, D.; Beaussire, L.; Gillibert, A.; Blanchard, F.; Toure, E.; Bazille, C.; Perdrix, A.; Ziegler, F.; Gangloff, A.; Hassine, M.; et al. CEA, CA19-9, circulating DNA and circulating tumour cell kinetics in patients treated for metastatic colorectal cancer (mCRC). Br. J. Cancer 2021, 125, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, C.; Weickmann, S.; Schmidt, B.; Fleischhacker, M. An improved method for the isolation of free-circulating plasma DNA and cell-free DNA from other body fluids. Ann. N. Y. Acad. Sci. 2008, 1137, 135–139. [Google Scholar] [CrossRef]
- Severson, E.A.; Riedlinger, G.M.; Connelly, C.F.; Vergilio, J.A.; Goldfinger, M.; Ramkissoon, S.; Frampton, G.M.; Ross, J.S.; Fratella-Calabrese, A.; Gay, L.; et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood 2018, 131, 2501–2505. [Google Scholar] [CrossRef] [Green Version]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal-Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.A.; Hachiya, T.; Iwaya, T.; Kume, K.; Matsuo, T.; Kawasaki, K.; Abiko, Y.; Akasaka, R.; Matsumoto, T.; Otsuka, K.; et al. Individualized Mutation Detection in Circulating Tumor DNA for Monitoring Colorectal Tumor Burden Using a Cancer-Associated Gene Sequencing Panel. PLoS ONE 2016, 11, e0146275. [Google Scholar] [CrossRef]
- Olmedillas-López, S.; García-Olmo, D.C.; García-Arranz, M.; Peiró-Pastor, R.; Aguado, B.; García-Olmo, D. Liquid biopsy by NGS: Differential presence of exons (DPE) in cell-free DNA reveals different patterns in metastatic and nonmetastatic colorectal cancer. Cancer Med. 2018, 7, 1706–1716. [Google Scholar] [CrossRef]
- Yang, Y.C.; Wang, D.; Jin, L.; Yao, H.W.; Zhang, J.H.; Wang, J.; Zhao, X.M.; Shen, C.Y.; Chen, W.; Wang, X.L.; et al. Circulating tumor DNA detectable in early- and late-stage colorectal cancer patients. Biosci. Rep. 2018, 38, BSR20180322. [Google Scholar] [CrossRef] [Green Version]
- Spindler, K.G.; Boysen, A.K.; Pallisgård, N.; Johansen, J.S.; Tabernero, J.; Sørensen, M.M.; Jensen, B.V.; Hansen, T.F.; Sefrioui, D.; Andersen, R.F.; et al. Cell-Free DNA in Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. Oncologist 2017, 22, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Hamfjord, J.; Guren, T.K.; Dajani, O.; Johansen, J.S.; Glimelius, B.; Sorbye, H.; Pfeiffer, P.; Lingjærde, O.C.; Tveit, K.M.; Kure, E.H.; et al. Total circulating cell-free DNA as a prognostic biomarker in metastatic colorectal cancer before first-line oxaliplatin-based chemotherapy. Ann. Oncol. 2019, 30, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.; Wang, Y.; Cohen, J.; Li, L.; Hong, W.; Christie, M.; Wong, H.L.; Kosmider, S.; Wong, R.; Thomson, B.; et al. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: A prospective cohort study. PLoS Med. 2021, 18, e1003620. [Google Scholar] [CrossRef] [PubMed]
- Øgaard, N.; Reinert, T.; Henriksen, T.V.; Frydendahl, A.; Aagaard, E.; Ørntoft, M.W.; Larsen, M.Ø.; Knudsen, A.R.; Mortensen, F.V.; Andersen, C.L. Tumour-agnostic circulating tumour DNA analysis for improved recurrence surveillance after resection of colorectal liver metastases: A prospective cohort study. Eur. J. Cancer 2022, 163, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Herbst, A.; Behrens, A.; Crispin, A.; Stieber, P.; Göke, B.; Lamerz, R.; Kolligs, F.T. Methylation of serum DNA is an independent prognostic marker in colorectal cancer. Clin. Cancer Res. 2006, 12, 7347–7352. [Google Scholar] [CrossRef] [Green Version]
- Young, G.P.; Symonds, E.L.; Nielsen, H.J.; Ferm, L.; Christensen, I.J.; Dekker, E.; van der Vlugt, M.; Mallant-Hent, R.C.; Boulter, N.; Yu, B.; et al. Evaluation of a panel of tumor-specific differentially-methylated DNA regions in IRF4, IKZF1 and BCAT1 for blood-based detection of colorectal cancer. Clin. Epigenetics 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Picardo, F.; Romanelli, A.; Muinelo-Romay, L.; Mazza, T.; Fusilli, C.; Parrella, P.; Barbazán, J.; Lopez-López, R.; Barbano, R.; De Robertis, M.; et al. Diagnostic and Prognostic Value of B4GALT1 Hypermethylation and Its Clinical Significance as a Novel Circulating Cell-Free DNA Biomarker in Colorectal Cancer. Cancers 2019, 11, 1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipp, A.B.; Stieber, P.; Nagel, D.; Neumann, J.; Spelsberg, F.; Jung, A.; Lamerz, R.; Herbst, A.; Kolligs, F.T. Prognostic role of methylated free circulating DNA in colorectal cancer. Int. J. Cancer 2012, 131, 2308–2319. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.L.; Lim, J.S.; Sinha, A.; Gopinathan, A.; Lim, R.; Tan, C.S.; Soh, T.; Venkatesh, S.; Titin, C.; Sapari, N.S.; et al. Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J. Transl. Med. 2015, 13, 57. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Lenz, H.J.; Siena, S.; Sobrero, A.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: A retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015, 16, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.A.L.; Lam, M.; Kanikarla-Marie, P.; Raghav, K.P.S.; Morris, V.K.; Brown, H.; Windham, J.; Duose, D.Y.; Overman, M.J.; Sanchez, E.V.; et al. Circulating tumor DNA (ctDNA) as an early marker to monitor clinical benefit of regorafenib and TAS-102 in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2018, 36 (Suppl. 15), 3533. [Google Scholar] [CrossRef]
- Colle, R.; Radzik, A.; Cohen, R.; Pellat, A.; Lopez-Tabada, D.; Cachanado, M.; Duval, A.; Svrcek, M.; Menu, Y.; André, T. Pseudoprogression in patients treated with immune checkpoint inhibitors for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer. Eur. J. Cancer 2021, 144, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated With Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chevalier, D.; Saluja, J.; Sandhu, J.; Lau, C.; Fakih, M. Regorafenib and Nivolumab or Pembrolizumab Combination and Circulating Tumor DNA Response Assessment in Refractory Microsatellite Stable Colorectal Cancer. Oncologist 2020, 25, e1188–e1194. [Google Scholar] [CrossRef]
- Raunkilde, L.; Hansen, T.F.; Andersen, R.F.; Havelund, B.M.; Thomsen, C.B.; Jensen, L.H. NPY Gene Methylation in Circulating Tumor DNA as an Early Biomarker for Treatment Effect in Metastatic Colorectal Cancer. Cancers 2022, 14, 4459. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Lenz, H.J.; Köhne, C.H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [Green Version]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.M.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. CAPRI-GOIM Investigators. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Satake, H.; Usher, J.; Jaimes, Y.; Miyamoto, Y.; Nakamura, M.; Kataoka, M.; Shiozawa, M.; Takagane, A.; Terazawa, T.; et al. Dynamic changes in RAS gene status in circulating tumour DNA: A phase II trial of first-line FOLFOXIRI plus bevacizumab for RAS-mutant metastatic colorectal cancer (JACCRO CC-11). ESMO Open 2022, 7, 100512. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Johnson, M.; Ou, S.; Barve, M.; Rybkin, I.; Papadopoulos, K.; Leal, T.; Velastegui, K.; Christensen, J.; Kheoh, T.; Chao, R.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in patients with Colorectal Cancer (CRC) and Other Solid Tumors Harboring a KRAS G12C Mutation. Eur. J. Cancer 2020, 138 (Suppl. 2), S2. [Google Scholar] [CrossRef]
- Thein, K.Z.; Biter, A.B.; Banks, K.C.; Duda, A.W.; Saam, J.; Roszik, J.; Janku, F.; Skoulidis, F.; Heymach, J.V.; Kopetz, S.; et al. Identification of KRASG12C Mutations in Circulating Tumor DNA in Patients With Cancer. JCO Precis. Oncol. 2022, 6, e2100547. [Google Scholar] [CrossRef]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.A.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.; Ou, F.S.; Venook, A.P.; Innocenti, F.; Sun, R.; Lenz, H.J.; Kopetz, S. Acquired Genomic Alterations on First-Line Chemotherapy With Cetuximab in Advanced Colorectal Cancer: Circulating Tumor DNA Analysis of the CALGB/SWOG-80405 Trial (Alliance). J. Clin. Oncol. 2023, 41, 472–478. [Google Scholar] [CrossRef]
- Topham, J.T.; O’Callaghan, C.J.; Feilotter, H.; Kennecke, H.F.; Lee, Y.S.; Li, W.; Banks, K.C.; Quinn, K.; Renouf, D.J.; Jonker, D.J.; et al. Circulating Tumor DNA Identifies Diverse Landscape of Acquired Resistance to Anti-Epidermal Growth Factor Receptor Therapy in Metastatic Colorectal Cancer. J. Clin. Oncol. 2023, 41, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parseghian, C.M.; Loree, J.M.; Morris, V.K.; Liu, X.; Clifton, K.K.; Napolitano, S.; Henry, J.T.; Pereira, A.A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciardiello, F.; Normanno, N.; Martinelli, E.; Troiani, T.; Pisconti, S.; Cardone, C.; Nappi, A.; Bordonaro, A.R.; Rachiglio, M.; Lambiase, M.; et al. CAPRI-GOIM investigators. Cetuximab continuation after first progression in metastatic colorectal cancer (CAPRI-GOIM): A randomized phase II trial of FOLFOX plus cetuximab versus FOLFOX. Ann. Oncol. 2016, 27, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Santini, D.; Vincenzi, B.; Addeo, R.; Garufi, C.; Masi, G.; Scartozzi, M.; Mancuso, A.; Frezza, A.M.; Venditti, O.; Imperatori, M.; et al. Cetuximab rechallenge in metastatic colorectal cancer patients: How to come away from acquired resistance? Ann. Oncol. 2012, 23, 2313–2318. [Google Scholar] [CrossRef]
- Masuishi, T.; Tsuji, A.; Kotaka, M.; Nakamura, M.; Kochi, M.; Takagane, A.; Shimada, K.; Denda, T.; Segawa, Y.; Tanioka, H.; et al. Phase 2 study of irinotecan plus cetuximab rechallenge as third-line treatment in KRAS wild-type metastatic colorectal cancer: JACCRO CC-08. Br. J. Cancer 2020, 123, 1490–1495. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients With RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol. 2021, 7, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Argilés, G.; Ciardiello, F.; Poulsen, T.T.; Dienstmann, R.; Kragh, M.; Kopetz, S.; Lindsted, T.; Ding, C.; Vidal, J.; et al. Efficacy of Sym004 in Patients With Metastatic Colorectal Cancer With Acquired Resistance to Anti-EGFR Therapy and Molecularly Selected by Circulating Tumor DNA Analyses: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2018, 4, e175245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Crisafulli, G.; Bartolini, A.; Fenocchio, E.; Amatu, A.; Manca, P.; et al. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: The phase 2 CHRONOS trial. Nat. Med. 2022, 28, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.; Virgili Manrique, A.C.; Capdevila, J.; Muñoz Boza, F.; Galván, P.; Richart, P.; Oliveres, H.; Páez, D.; Hernando, J.; Serrano, S.; et al. Randomized phase II trial of FOLFIRI-panitumumab compared with FOLFIRI alone in patients with RAS wild-type circulating tumor DNA metastatic colorectal cancer beyond progression to first-line FOLFOX-panitumumab: The BEYOND study (GEMCAD 17-01). Clin. Transl. Oncol. 2022, 24, 2155–2165. [Google Scholar] [CrossRef]
- Cicek, M.S.; Lindor, N.M.; Gallinger, S.; Bapat, B.; Hopper, J.L.; Jenkins, M.A.; Young, J.; Buchanan, D.; Walsh, M.D.; Le Marchand, L.; et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J. Mol. Diagn. 2011, 13, 271–281. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Raut, C.P.; Rodriguez-Bigas, M.A. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis. Markers 2004, 20, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, J.W.; Flanagan, L.A.; Smalley, R.L.; Nassif, N.A.; Burgart, L.J.; Halberg, R.B.; Megid, W.M.; Thibodeau, S.N. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis. Markers 2004, 20, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Silveira, A.B.; Bidard, F.C.; Kasperek, A.; Melaabi, S.; Tanguy, M.L.; Rodrigues, M.; Bataillon, G.; Cabel, L.; Buecher, B.; Pierga, J.Y.; et al. High-Accuracy Determination of Microsatellite Instability Compatible with Liquid Biopsies. Clin. Chem. 2020, 66, 606–613. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, W.H.; Kwak, Y.; Koh, J.; Bae, J.M.; Kim, K.M.; Chang, M.S.; Han, H.S.; Kim, J.M.; Kim, H.W.; et al. Gastrointestinal Pathology Study Group of Korean Society of Pathologists, & Molecular Pathology Study Group of Korean Society of Pathologists. Molecular Testing for Gastrointestinal Cancer. J. Pathol. Transl. Med. 2017, 51, 103–121. [Google Scholar] [PubMed] [Green Version]
- Ladas, I.; Yu, F.; Leong, K.W.; Fitarelli-Kiehl, M.; Song, C.; Ashtaputre, R.; Kulke, M.; Mamon, H.; Makrigiorgos, G.M. Enhanced detection of microsatellite instability using pre-PCR elimination of wild-type DNA homo-polymers in tissue and liquid biopsies. Nucleic Acids Res. 2018, 46, e74. [Google Scholar] [CrossRef] [Green Version]
- Trabucco, S.E.; Gowen, K.; Maund, S.L.; Sanford, E.; Fabrizio, D.A.; Hall, M.J.; Yakirevich, E.; Gregg, J.P.; Stephens, P.J.; Frampton, G.M.; et al. A Novel Next-Generation Sequencing Approach to Detecting Microsatellite Instability and Pan-Tumor Characterization of 1000 Microsatellite Instability-High Cases in 67,000 Patient Samples. J. Mol. Diagn. 2019, 21, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Mazzaferro, V.; Miceli, R.; Cotsoglou, C.; Melotti, F.; Fanetti, G.; Perrone, F.; Biondani, P.; Muscarà, C.; Di Bartolomeo, M.; et al. Pathological response after neoadjuvant bevacizumab- or cetuximab-based chemotherapy in resected colorectal cancer liver metastases. Med. Oncol. 2015, 32, 182. [Google Scholar] [CrossRef]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; André, T.; Yoshino, T.; Bendell, J.C.; Atreya, C.E.; Schellens, J.H.M.; Ducreux, M.P.; McRee, A.; Siena, S.; Middleton, G.; et al. Efficacy and circulating tumor DNA (ctDNA) analysis of the BRAF inhibitor dabrafenib (D), MEK inhibitor trametinib (T), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E–mutated (BRAFm) metastatic colorectal cancer (mCRC). Ann. Oncol. 2016, 27, vi150. [Google Scholar] [CrossRef]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.F.; Huang, Z.Y.; Chen, X.X.; Chen, Z.G.; Wu, S.X.; Ren, C.; Hu, M.T.; Bao, H.; Jin, Y.; Wang, F.; et al. Monitoring tumour resistance to the BRAF inhibitor combination regimen in colorectal cancer patients via circulating tumour DNA. Drug Resist. Updates 2022, 65, 100883. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Gao, H.; Guan, M.; Sun, Z.; Bai, C. High c-Met expression is a negative prognostic marker for colorectal cancer: A meta-analysis. Tumour Biol. 2015, 36, 515–520. [Google Scholar] [CrossRef]
- Raghav, K.; Morris, V.; Tang, C.; Morelli, P.; Amin, H.M.; Chen, K.; Manyam, G.C.; Broom, B.; Overman, M.J.; Shaw, K.; et al. MET amplification in metastatic colorectal cancer: An acquired response to EGFR inhibition, not a de novo phenomenon. Oncotarget 2016, 7, 54627–54631. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Delord, J.P.; Argilés, G.; Fayette, J.; Wirth, L.; Kasper, S.; Siena, S.; Mesia, R.; Berardi, R.; Cervantes, A.; Dekervel, J.; et al. A phase 1b study of the MET inhibitor capmatinib combined with cetuximab in patients with MET-positive colorectal cancer who had progressed following anti-EGFR monoclonal antibody treatment. Investig. New Drugs 2020, 38, 1774–1783. [Google Scholar] [CrossRef]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Magliocco, A.M.; Kim, J.; Okamoto, W.; Kim, J.E.; Sawada, K.; Nakamura, Y.; Kopetz, S.; Park, W.Y.; Tsuchihara, K.; et al. International Harmonization of Provisional Diagnostic Criteria for ERBB2-Amplified Metastatic Colorectal Cancer Allowing for Screening by Next-Generation Sequencing Panel. JCO Precis. Oncol. 2020, 4, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Sartore-Bianchi, A.; Nagy, R.J.; Raghav, K.; Odegaard, J.I.; Lanman, R.B.; Trusolino, L.; Marsoni, S.; Siena, S.; Bardelli, A. Plasma HER2 (ERBB2) Copy Number Predicts Response to HER2-targeted Therapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 3046–3053. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.; Mileyko, V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Lo, Y.M.D. The Long and Short of Circulating Cell-Free DNA and the Ins and Outs of Molecular Diagnostics. Trends Genet. TIG 2016, 32, 360–371. [Google Scholar] [CrossRef]
- Jiang, P.; Sun, K.; Peng, W.; Cheng, S.H.; Ni, M.; Yeung, P.C.; Heung, M.M.S.; Xie, T.; Shang, H.; Zhou, Z.; et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020, 10, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girirajan, S.; Campbell, C.; Eichler, E. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Physiol. Behav. 2016, 176, 139–148. [Google Scholar]
- Mathios, D.; Johansen, J.S.; Cristiano, S.; Medina, J.E.; Phallen, J.; Larsen, K.R.; Bruhm, D.C.; Niknafs, N.; Ferreira, L.; Adleff, V.; et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021, 12, 5060. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Wang, Z.; Ma, X.; Guo, W.; Zhang, X.; Tang, W.; Chen, X.; Wang, X.; Chen, Y.; Mo, S.; et al. Letter to the Editor: An ultra-sensitive assay using cell-free DNA fragmentomics for multi-cancer early detection. Mol. Cancer 2022, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, Y.; Tang, W.; Bao, H.; Mo, S.; Liu, R.; Wu, S.; Bao, H.; Li, Y.; Zhang, L.; et al. Multi-dimensional fragmentomic assay for ultrasensitive early detection of colorectal advanced adenoma and adenocarcinoma. J. Hematol. Oncol. 2021, 14, 175. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Bao, H.; Xia, F.; Wan, J.; Shen, L.; Wang, Y.; Zhang, H.; Wei, Y.; Wu, X.; et al. Utility of Circulating Free DNA Fragmentomics in the Prediction of Pathological Response after Neoadjuvant Chemoradiotherapy in Locally Advanced Rectal Cancer. Clin Chem. 2023, 69, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Roch, B.; Mazard, T.; Blache, P.; Dache, Z.A.A.; Pastor, B.; Pisareva, E.; Tanos, R.; Thierry, A.R. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021, 6, e144561. [Google Scholar] [CrossRef] [PubMed]
- Van’t Erve, I.; Rovers, K.P.; Constantinides, A.; Bolhuis, K.; Wassenaar, E.C.; Lurvink, R.J.; Huysentruyt, C.J.; Snaebjornsson, P.; Boerma, D.; van den Broek, D.; et al. Detection of tumor-derived cell-free DNA from colorectal cancer peritoneal metastases in plasma and peritoneal fluid. J. Pathol. Clin. Res. 2021, 7, 203–208. [Google Scholar] [CrossRef]
- Nicolazzo, C.; Belardinilli, F.; Vestri, A.; Magri, V.; De Renzi, G.; De Meo, M.; Caponnetto, S.; Di Nicolantonio, F.; Cortesi, E.; Giannini, G.; et al. RAS Mutation Conversion in Bevacizumab-Treated Metastatic Colorectal Cancer Patients: A Liquid Biopsy Based Study. Cancers 2022, 14, 802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Exosomal | Molecule Type | Sample Type | Detection Technique | Trend | Application |
|---|---|---|---|---|---|
| QSOX1 | protein | plasma | ultracentrifugation | downregulated | diagnostic |
| Copine3 | protein | plasma | ELISA | upregulated | diagnostic prognostic |
| miR-21 | miRNA | plasma | ultracentrifugation | upregulated | diagnostic prognostic |
| miR-122 | miRNA | serum | Invitrogen Exosome Isolation Kit | upregulated | diagnostic prognostic |
| miR-25-3p | miRNA | serum | ultracentrifugation | upregulated | prognostic |
| miR-28-3p let-7e-5p miR-106a-5p miR-542-5p | miRNA | plasma | TaqMan Low-Density Array | upregulated | diagnostic |
| KRAS mut | ExoDNA | plasma | ultracentrifugation ddPCR | upregulated | diagnostic prognostic predictive |
| UCA1 | lncRNAs | serum | ultracentrifugation ExoQuick | downregulated | diagnostic |
| CRNDE-h | lncRNAs | serum | ExoQuick | upregulated | diagnostic prognostic |
| miR-17-92a | miRNA | serum | ultracentrifugation | upregulated | prognostic |
| miR-320d | miRNA | serum | ultracentrifugation | upregulated | diagnostic |
| miR-203 | miRNA | serum | ultracentrifugation | upregulated | prognostic |
| miR-17-5p miR-92a-3p | miRNA | serum | ultracentrifugation | upregulated | diagnostic |
| hsa-circ0004771 | circRNA | serum | Invitrogen Exosome Isolation Kit | upregulated | diagnostic prognostic |
| miR-196b-5p | miRNA | serum | exoRNeasy Kit | upregulated | predictive (5-fluorouracil resistance) |
| miR-21-5p miR-1246 miR-1229-5p miR-96-5p | miRNA | serum | ultracentrifugation | upregulated | predictive (oxaliplatin/5-fluorouracil resistance) |
| miR-222 | miRNA | serum | ultracentrifugation | upregulated | diagnostic prognostic |
| UCA1 | lncRNAs | serum | ultracentrifugation ExoQuick | Upregulated | predictive (cetuximab resistance) |
| Trial Name and Author | Study Type | Rechallenge Treatment | Line | N | OS | PFS | ORR | ctDNA Selection |
|---|---|---|---|---|---|---|---|---|
| Santini et al. | Retrospective | Irinotecan + cetuximab | ≥3rd | 39 | NR | 6.6 m | 53.8% | None |
| Liu et al. | Retrospective | Cetuximab ± erlotinib | ≥2nd | 89 | NR | 4.9 m for prior responder vs. 2.5 m for prior no-responder (p = 0.064) | NR | None |
| Tanioka et al. | Retrospective | Irinotecan + cetuximab | ≥3rd | 14 | NR | 4.4 m | 21.4% | None |
| Rossini et al. | Retrospective | FOLFIRI + cetuximab/FOLFOX + panitumumab/CapIri + cetuximab/Irinotecan + panitumumab/Irinotecan + cetuximab/Cetuximab/Panitumumab | ≥3rd | 86 | 10.2 m | 3.8 m | 19.8% | None |
| Karani et al. | Retrospective | Cetuximab ± CT | ≥3rd | 17 | 7.5 m | 3.3 m | 18% | None |
| Chong et al. | Retrospective | Anti-EGFR ± CT | ≥2nd | 22 | 7.7 m | 4.1 m | 4.5% | None |
| CAPRI-GOIM (Ciardello et al.) | Prospective | FOLFOX + cetuximab vs. FOLFOX (PD after FOLFIRI + cetuximab in WT pt) | 2nd | 66 (pt WT in retrospective NGS analysis) | 23.7 m vs. 19.8 m (HR 0.57, 95% CI 0.32–1.02; p = 0.056) | 6.9 m vs. 5.3 m (HR 0.56, 95% CI 0.33–0.94; p = 0.025) | 29.4% vs. 9.4% | None |
| CRICKET (Cremolini et al.) | Prospective | Irinotecan + cetuximab | 3rd | 28 | 9.8 m (12.5 m for ctDNA WT vs. 5.2 m for ctDNA M; HR 0.58, 95% CI 0.22–1.52; p = 0.24) | 3.4 m (4 m for ctDNA WT vs. 1.9 m for ctDNA M;HR 0.44, 95% CI 0.18–0.98; p = 0.03) | 14% (all ctDNA WT) | Retrospective analysis |
| PACER (Piccirillo et al.) | Prospective | Panitumumab | ≥2nd | 41 (all WT before rechallenge) | 6.8 m | 2.1 m | 7.3% | None |
| JACCRO CC-08 (Masuishi et al.) | Prospective | Irinotecan + cetuximab | 3rd | 34 (all baseline WT) | 8.2 m | 2.4 m | 2.9% | None |
| JACCRO CC-09 (Tsuji et al.) | Prospective | Irinotecan + panitumumab | 3rd | 25 (all baseline WT) | 8.9 m | 3.1 m | 8.3% | None |
| Sunakawa et al. | Retrospective(post-hoc analysis of JACCRO CC-08 and CC-09) | Irinotecan + anti-EGFR | 3rd | 16 (all baseline WT) | 8.9 m (3.8 m for ctDNA M vs. 16 m for ctDNA WT; HR 12.4, 95% CI 2.7–87.7; p = 0.0028) | 3.1 m (2.3 m for ctDNA M vs. 4.7 m for ctDNA WT; HR 6.2, 95% CI 1.6–30.5; p = 0.013) | 0% | Retrospective analysis |
| CAVE (Martinelli et al.) | Prospective | Avelumab + cetuximab | 3rd | 77 (all baseline WT) | 11.6 m (17.3m for ctDNA WT vs. 10.4 m for ctDNA M; HR 0.49, 95% CI 0.27–0.90; p = 0.02) | 3.6 m (4.1 m for ctDNA WT vs. 3 m for ctDNA M;HR 0.42, 95% CI 0.23–0.75; p = 0.004) | 7.8% (8.5% for WT vs. 5.1% for M) | Retrospective analysis |
| BEYOND (Aparicio et al.) | Prospective | FOLFIRI + panitumumab vs. FOLFIRI | 2nd | 31 (all WT ctDNA before retreatment) | 13 m vs. 10 m (HR 0.55, 95% CI 0.2–1.48) | 11 m vs. 4 m (HR 0.58, 95% CI 0.25–1.3) | 33% vs. 7.7% | Interventional |
| CHRONOS (Sartore-Bianchi et al.) | Prospective | Panitumumab | ≥3rd | 27 (all WT ctDNA before rechallenge) | 55 wks | 16.4 wks | 30% | Interventional |
| E-RECHALLANGE (Nakamura et al.) | Prospective | Irinotecan + cetuximab | ≥3rd | 33 (all WT at baseline) | 8.6 m | 2.9 m (7 m for ctDNA WT vs. 2.9 m for ctDNA M) | 15.6% (50% in ctDNA WT) | Retrospective analysis |
| Montagut et al. | Prospective | Sym004 (futuximab + modotuximab) 12 mg/Kg (arm A) vs. Sym004 6 mg/Kg (arm B) vs. SoC (arm C) | ≥3rd | 254 (all WT at baseline and acquired resistance to prior anti-EGFR therapy) | 7.9 m vs. 10.3 m vs. 9.6 m (HR 1.31, 95% CI 0.92–1.87 for A vs. C; HR 0.97, 95% CI 0.68–1.4 for B vs. C) In ctDNA WT: 10.6m vs. 12.8m vs. 7.3m | 2.8 m vs. 2.7 m vs. 2.6 m | 14.1% vs. 9.6% vs. 2.9% | Retrospective analysis |
| Mariani et al. | Retrospective | Irinotecan + cetuximab or cetuximab | ≥3rd | 26 (all WT ctDNA before rechallenge) | 5 m | 3.5 m | 25% | Retrospective analysis |
| D’Onofrio et al. | Prospective | CT + anti-EGFR | ≥3rd | 10 (all WT ctDNA before rechallenge) | NR | 11.3 m | 70% | Interventional |
| Name (NCT Number) | Phase | Setting | Line | Treatment Arms | N (Actual/Estimated Enrollment) | Primary Endpoints | Status |
|---|---|---|---|---|---|---|---|
| PULSE (NCT03992456) | 2 | Rechallenge | 3rd | Panitumumab vs. regorafenib or trifluridine/tipiracil | 120 | OS | Active, not recruiting |
| PARERE (NCT04787341) | 2 | Rechallenge | 3rd–4th (sequence strategy) | Panitumumab followed by regorafenib vs. regorafenib followed by panitumumab | 214 | OS | Recruiting |
| CAPRI 2 GOIM (NCT05312398) | 2 | Rechallenge/reintroduction | 2nd–3rd (1L FOLFIRI + Cetuximab) | 2L FOLFOX + cetuximab (ctDNA WT) or FOLFOX + bevacizumab (ctDNA M) 3L irinotecan + cetuximab (ctDNA WT) vs. regorafenib or trifluridine/tipiracil (ctDNA M) | 200 | ORR | Recruiting |
| NCT04775862 | 2 | Rechallenge | 3rd | Anti-EGFR (ctDNA WT) or SoC (ctDNA M) | 60 | ORR and PFS | Recruiting |
| CITRIC (EudraCT Number:2020-000443-31) | 2 | Rechallenge | 3rd | Irinotecan + cetuximab vs. regorafenib or trifluridine/tipiracil | 66 | ORR | Recruiting |
| PURSUIT (jRCTs031190096) | 2 | Rechallenge | 3rd | Irinotecan + panitumumab | 50 | ORR | Not yet recruiting |
| NCT04509635 | 3 | Rechallenge | 3rd (non-resectable liver metastases) | Cetuximab + CT vs. CT | 50 | DCR | Not yet recruiting |
| NCT03844620 | 2 | Response to treatment | ≥3rd | Regorafenib or trifluridine/tipiracil | 100 | Early change in ctDNA as a predictor of radiological progression; safety | Recruiting |
| NCT04831528 | 2 | Target therapy | Progression at cetuximab-based therapy | 2L target therapy according to ctDNA analysis | 100 | ORR | Not yet recruiting |
| FOLICOLOR (NCT04735900) | NA | Response to treatment | 1 L (WT) | FOLFOX/FOLFIRI + panitumumab | 60 | To evaluate response and progression by NPY methylation(ctDNA) | Recruiting |
| LIBImAb (NCT04776655) | 3 | Efficacy of treatment | 1 L (WT on solid tumor biopsy but M at liquid biopsy) | Bevacizumab + FOLFIRI vs. cetuximab + FOLFIRI | 280 | PFS | Recruiting |
| COPERNIC (NCT05487248) | NA | Response to treatment | ≥3rd | SoC | 103 | To select timepoint and cut-off value for early on-treatment ctDNA changes | Not yet recruiting |
| OPTIMISE (NCT04680260) | 2 | Treatment selection and follow up | Oligometastatic CRC treated with local therapy (escalation or de-escalation CT/observation) | ctDNA-guided treatment approach vs. SoC | 350 | Recurrence-free rate | Recruiting |
| NCT05495672 | NA | Treatment selection and follow up | mCRC with metastatic small pulmonary nodules (local therapy or observation) | ctDNA-guided treatment approach | 100 | PFS | Recruiting |
| NCT03436563, cohort D | 1b/2 | Treatment selection | Oligometastatic CRC MSI with positive ctDNA following resection of liver metastases | Anti-PD-L1/TGFbetaRII fusion protein M7824 | NA | Clearance ctDNA | Active, not recruiting |
| NCT04555369 | NA | Response to treatment | mCRC receiving CT | ctDNA testing | 300 | ORR | Recruiting |
| NCT05141721 | 2/3 | Response to treatment | Maintenance therapy in mCRC pt after SoC | GRT-C901/GRT-R902 (neoantigen vaccine) + ipilimumab + atezolizumab + fluoropyrimidine + bevacizumab vs. fluoropyrimidine + bevacizumab | 665 | Antitumor activity by number of pt with ≥50% decrease from baseline in ctDNA; PFS | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delcuratolo, M.D.; Modrego-Sánchez, A.; Bungaro, M.; Antón-Pascual, B.; Teran, S.; Dipace, V.; Novello, S.; Garcia-Carbonero, R.; Passiglia, F.; Graválos-Castro, C. Liquid Biopsy in Advanced Colorectal Cancer: Clinical Applications of Different Analytes. J. Mol. Pathol. 2023, 4, 128-155. https://doi.org/10.3390/jmp4030013
Delcuratolo MD, Modrego-Sánchez A, Bungaro M, Antón-Pascual B, Teran S, Dipace V, Novello S, Garcia-Carbonero R, Passiglia F, Graválos-Castro C. Liquid Biopsy in Advanced Colorectal Cancer: Clinical Applications of Different Analytes. Journal of Molecular Pathology. 2023; 4(3):128-155. https://doi.org/10.3390/jmp4030013
Chicago/Turabian StyleDelcuratolo, Marco Donatello, Andrea Modrego-Sánchez, Maristella Bungaro, Beatriz Antón-Pascual, Santiago Teran, Valentina Dipace, Silvia Novello, Rocio Garcia-Carbonero, Francesco Passiglia, and Cristina Graválos-Castro. 2023. "Liquid Biopsy in Advanced Colorectal Cancer: Clinical Applications of Different Analytes" Journal of Molecular Pathology 4, no. 3: 128-155. https://doi.org/10.3390/jmp4030013