2. Results and Discussion
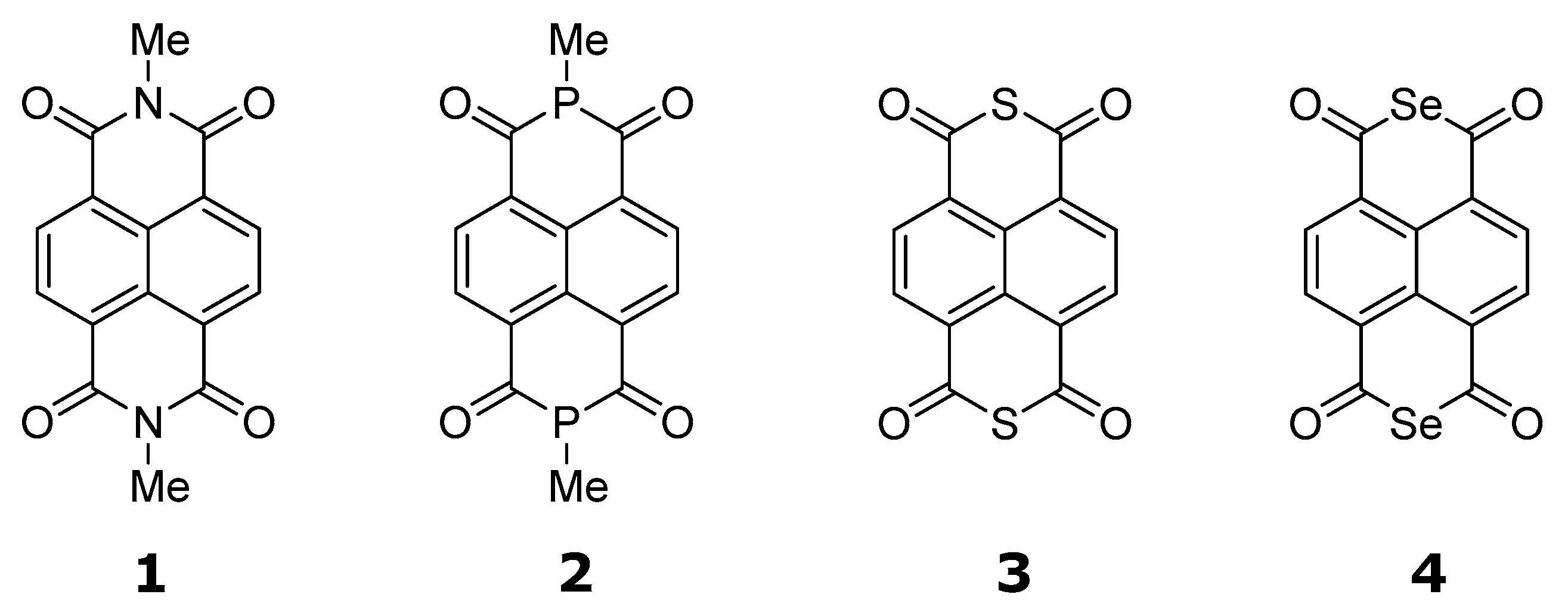
As model compounds for the study, the following compounds were chosen: 2,7-dimethylbenzo[
lmn][3]phenanthroline-1,3,6,8(2H,7H)-tetraone (
1), 2,7-dimethylisophosphinolino[6,5,4-
def]isophosphinoline-1,3,6,8(2H,7H)-tetraone (
2), isothiochromeno[6,5,4-def]isothiochromene-1,3,6,8-tetraone (
3), and isoselenochromeno[6,5,4-def]isoselenochromene-1,3,6,8-tetraone (
4) (
Figure 1).
In the case of
1 and
2, the positions 2 and 7 of the pyrene-1,3,6,8(2H,7H)-tetraone skeleton were occupied by N-Me and P-Me, to avoid the issue of the anion on the nitrogen or phosphorous atom, respectively, if the N-H or P-H were used [
5].
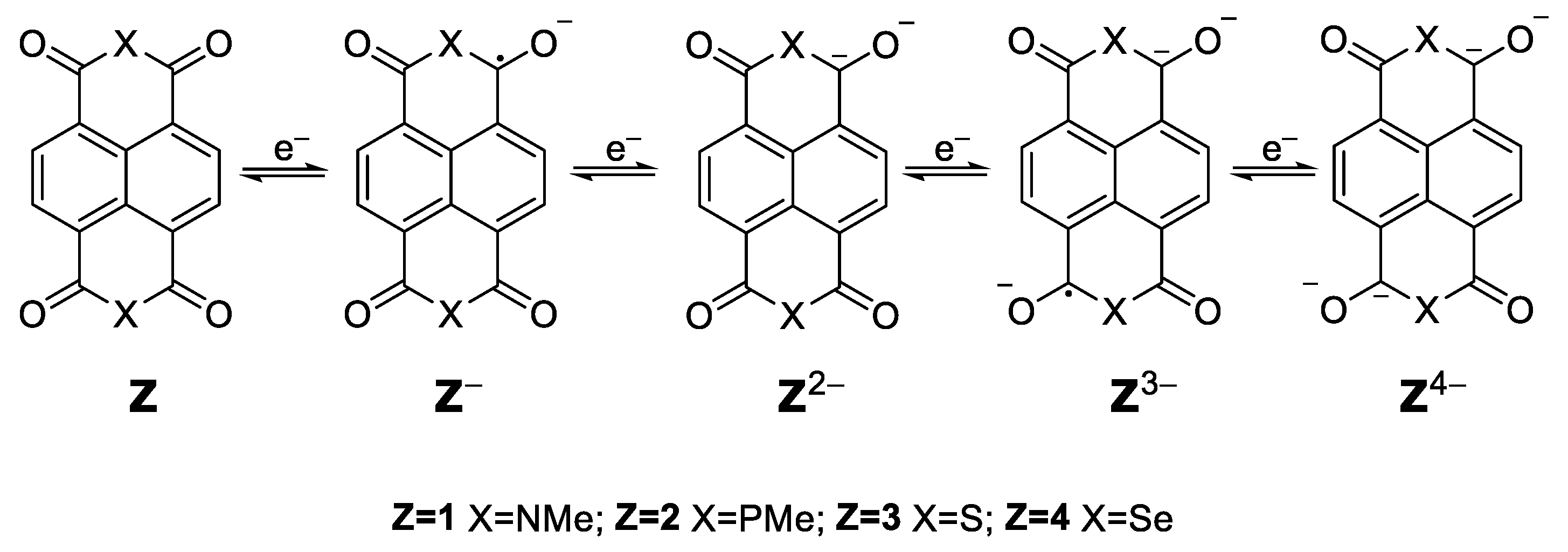
The process can be generalized as the enolization and its reverse process in the carbonyl group, which can be promoted by a conjugated structure, either aromatic or aliphatic [
3]. When a compound is used as the cathode material for rechargeable lithium batteries, its reduction and oxidation are accompanied by the association and disassociation of Li
+ ions with oxygen. Ideally, each formula unit is able to transfer up to four electrons through four consecutive steps (
Scheme 1).
Electron transfer during an electrochemical process leads to the redox of the compound. The redox ability of the compound can be quantitatively described by the redox potential. The performance of organic electrical devices is highly dependent on the oxidation potential () and the reduction potential () of the used materials. These potentials for the materials govern their capability to capture (or inject) holes and electrons, respectively, in the devices.
The thermodynamic cycle for the Gibbs free energy of the oxidation and reduction reaction of the molecule A is displayed in
Scheme 2 [
6].
The is evaluated as the electronic energy difference of the molecule in the gas phase and the solvated one using the equilibrium geometry obtained in vacuum.
Marcus theory was used to estimate the charge transfer properties. According to this theory, the reorganization energy (
) has both intra- and intermolecular contributions. The former reflects the deformation of molecular geometry in order to accommodate charge transfer; and the latter reflects the electronic polarization of the surrounding molecules, being much smaller than the intramolecular one and is usually neglected. The intramolecular reorganization energy can be evaluated either from the adiabatic potential-energy surfaces or from normal-mode analysis [
7]. In this method, the hole and electron reorganization energies (
) are defined by the following equation:
where,
and
are the ground-state energies of the optimized neutral and ionic states, respectively,
is the energy of the charged molecules at the optimal geometry of the neutral molecules, and
is the energy of the neutral molecules at the optimal ionic geometry.
The adiabatic ionization potential
and adiabatic electron affinity
are two important parameters to evaluate the oxidation and reduction ability of charged organic molecules. Especially, the large
is beneficial for stabilizing the organic radical anions. Meanwhile, the large
decreases the electron injection energy barrier and, hence, is helpful for electron transport [
8]. Thus, the corresponding adiabatic
s and
s were obtained with following equations:
All these calculations were carried out with Gaussian16 at B3LYP/6311g++(d,p) level [
9]. Dimethylsulfoxide (DMSO) was chosen (using a polarizable continuum model) for the solution calculations, due to its low toxicity and it is a non-hazardous solvent that can solubilize a vast variety of organic compounds [
10]. The vibrational frequency analysis was performed at the same level of theory and the resulted positive frequencies confirmed that the optimized geometries were found at the real minima on the potential energy surfaces.
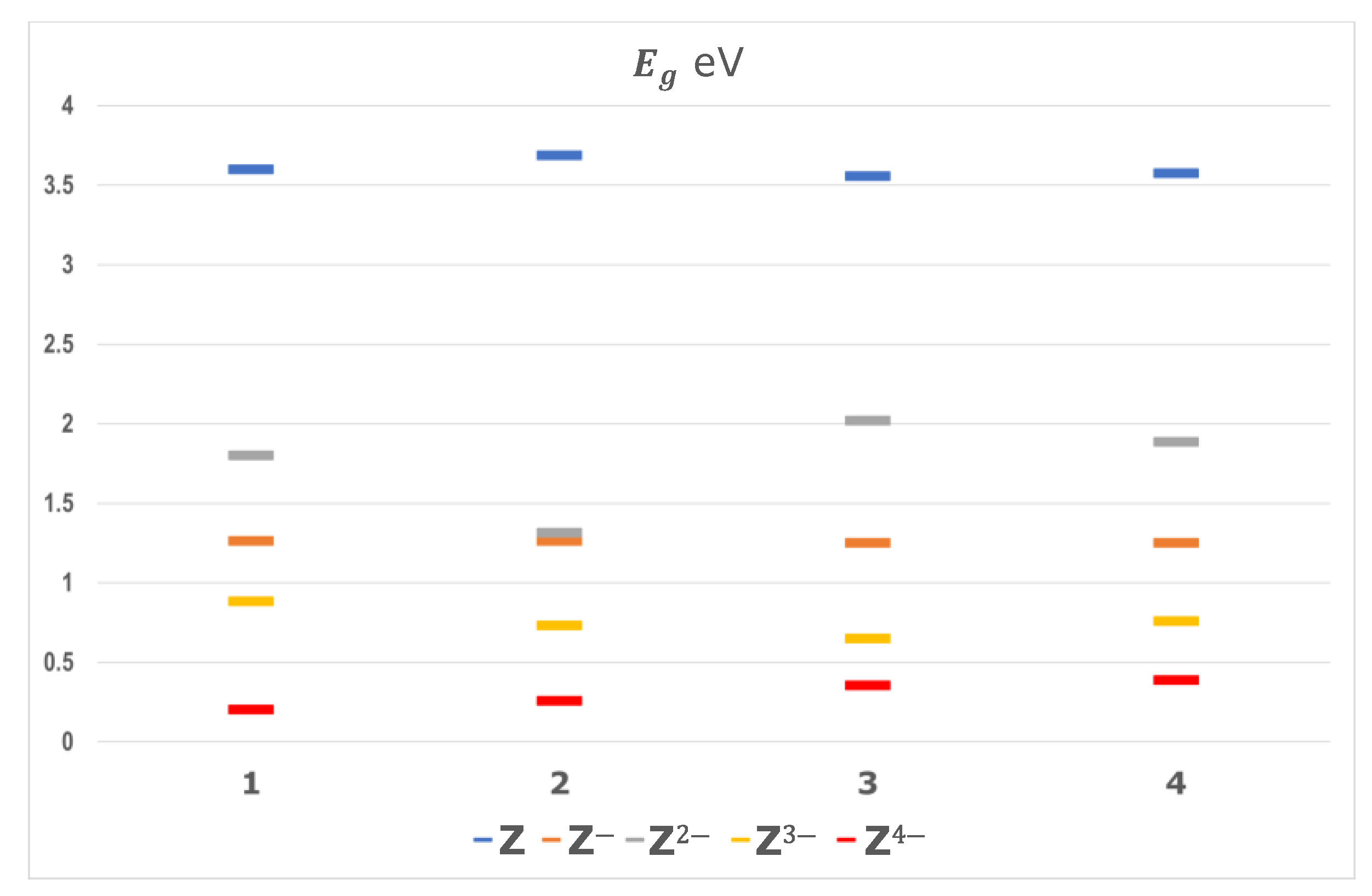
The molecular structure of designed molecules in their ground state was fully optimized and their optimized geometries rendered the energy and shape of their LUMO and HOMO. This was carried out for each of the species involved in the four steps of the electronic transference between the neutral state and the tetravalent anion. The energy of the frontier molecular orbitals of organic materials is one of the key factors in influencing the charge transport properties and the intrinsic air-stability of the materials. The energy gap (
) for the five states of the compounds follows the order
, the higher values being for the neutral species (
Figure 2).
Molecular electrostatic potential surfaces enable the visualization of the charge distributions and the charge-related properties of the molecules. Electronic total density and electrostatic cube files were generated for each species and Gabedit 2.5.1 [
11] was used to create the electrostatic potential mapped on the electronic density surface graphics shown in
Table 1. The colour code is between −0.4 and 0.1 eV for all examples. Carbonyl oxygens are the most negative areas in the surfaces.
The redox potentials for [Z]
0/−, [Z]
0/2−, [Z]
0/3−, [Z]
0/4− (
Scheme 3) were calculated as mentioned above, together with the corresponding electron reorganization energies (
λ) to check the feasibility of the reduction process. Lower
λ values are related to higher charge carrier mobility.
The reduction potentials (
Table 2) were higher with the increase in the number of electrons transferred, being for compounds
4 and
3 the larger ones. It is noteworthy that [
1]
0/− > [
2]
0/− and [
1]
0/2− > [
2]
0/2− but [
1]
0/3− < [
2]
0/3− and [
1]
0/4− < [
2]
0/4−. The electron reorganization energies show the higher value for the phosphorous compound, except for
2λ0/3−.
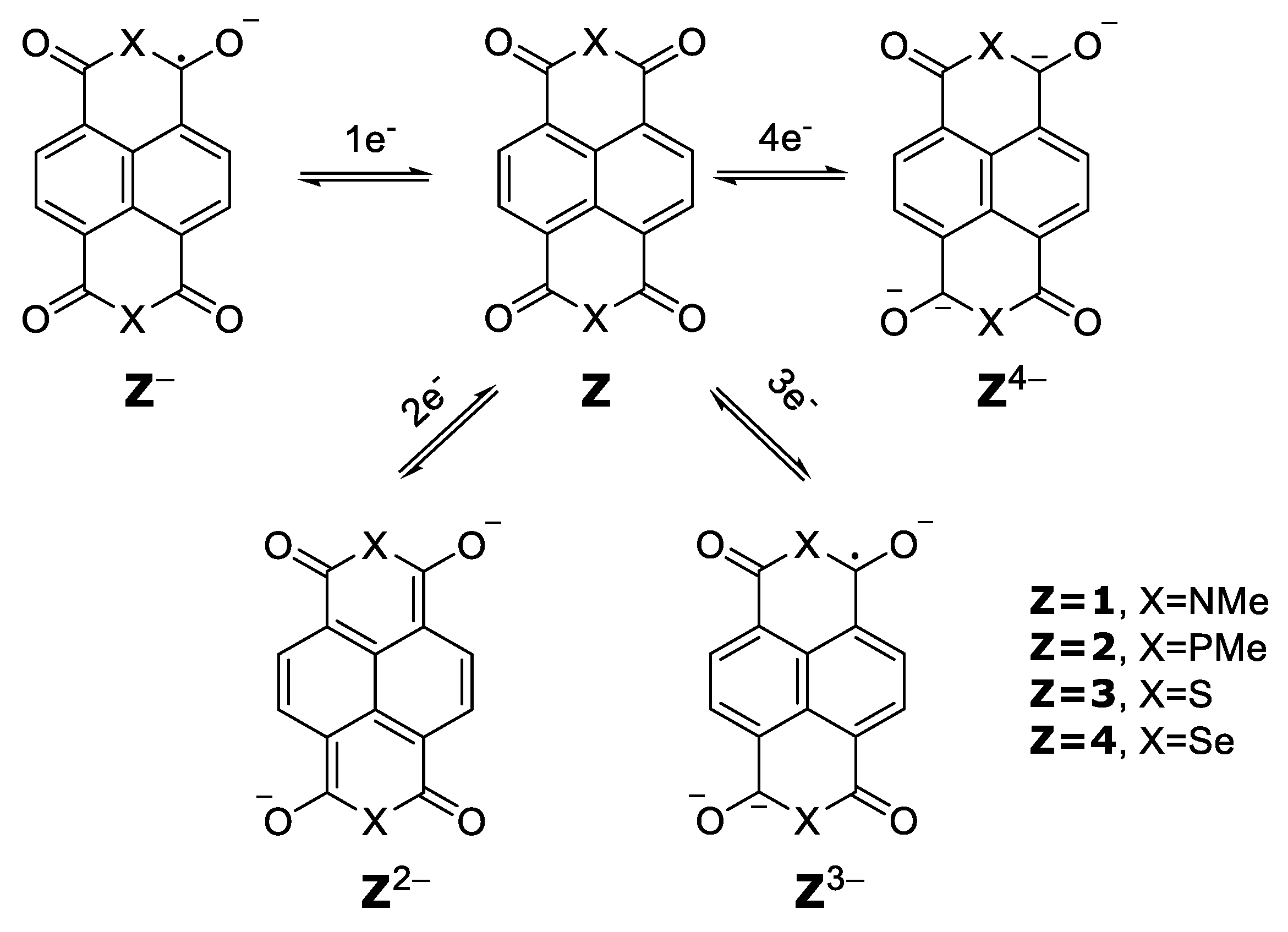
Reduction potentials for species [Z]
0/−, [Z
−]
−/2−, [Z
2−]
2−/3−, [Z
3−]
3−/4− (
Scheme 1), and their respective
λ values were calculated, in order to detect any anomalous value, which would evidence a special difficulty in the charge carrier mobility (
Table 3).
This approach to the evaluation of the reduction potentials turned out to be similar to the ones in
Table 2, with the potentials of the transfer of
1,
2,
3, and
4 electrons like the sum of the individual transfer.
Moreover, similar values are obtained for the reduction potentials of the compounds [
Z]
0/4−, both considering the joint transfer of the four electrons and the sum of the rearrangement energies of the four consecutive transfer processes of a single electron (
Table 4).
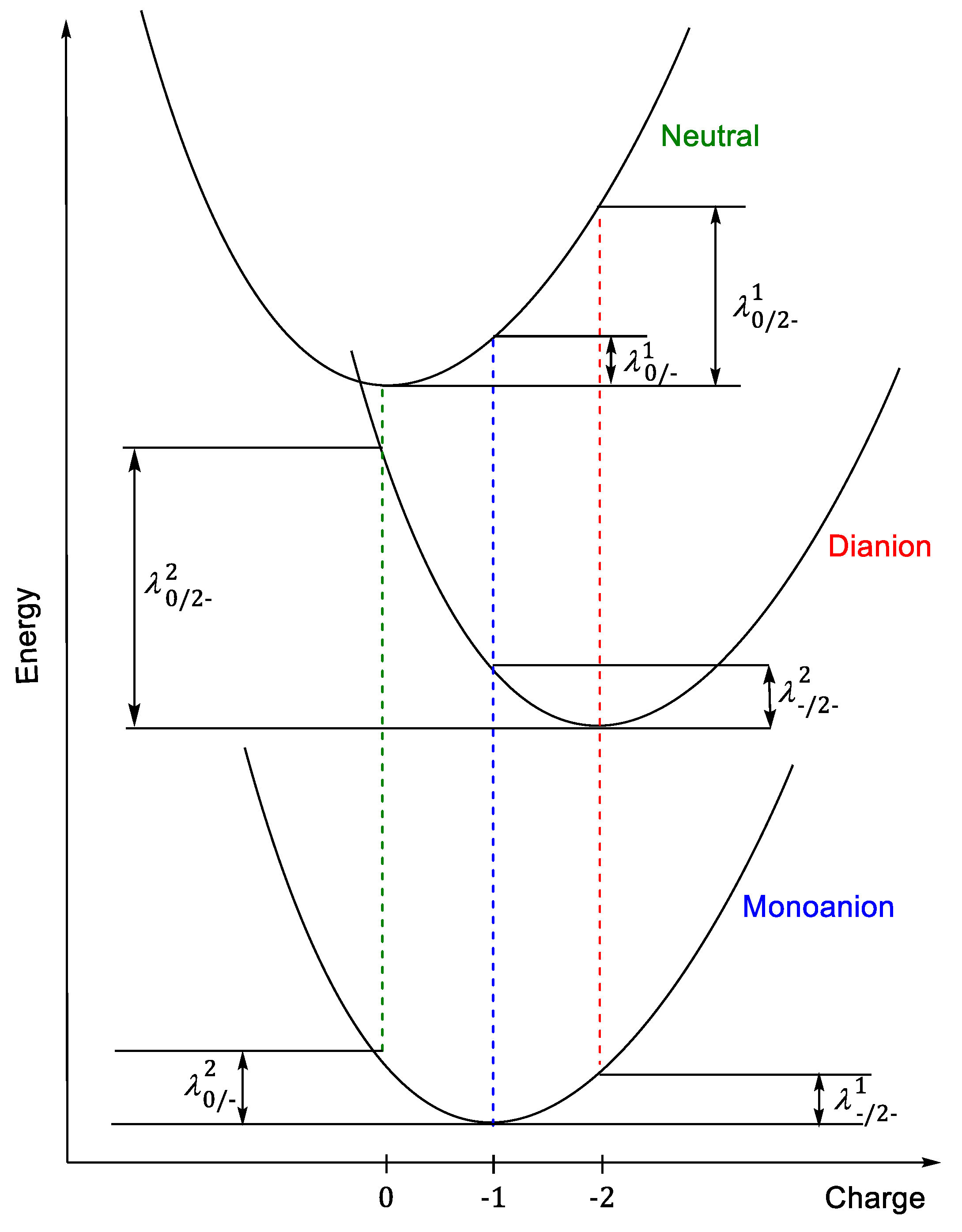
In evaluating electron rearrangement energy, the two alternatives are that either the transfer occurs with the motion of one electron, or it occurs simultaneously with several electrons at the same time [
12]. The adiabatic potential energy surface method, which is used for calculating the reorganization energy, is described in
Figure 3 for dianion for both different cases: through the monoanion with transfer of one electron in two stages and when two electrons are transferred in a single process.
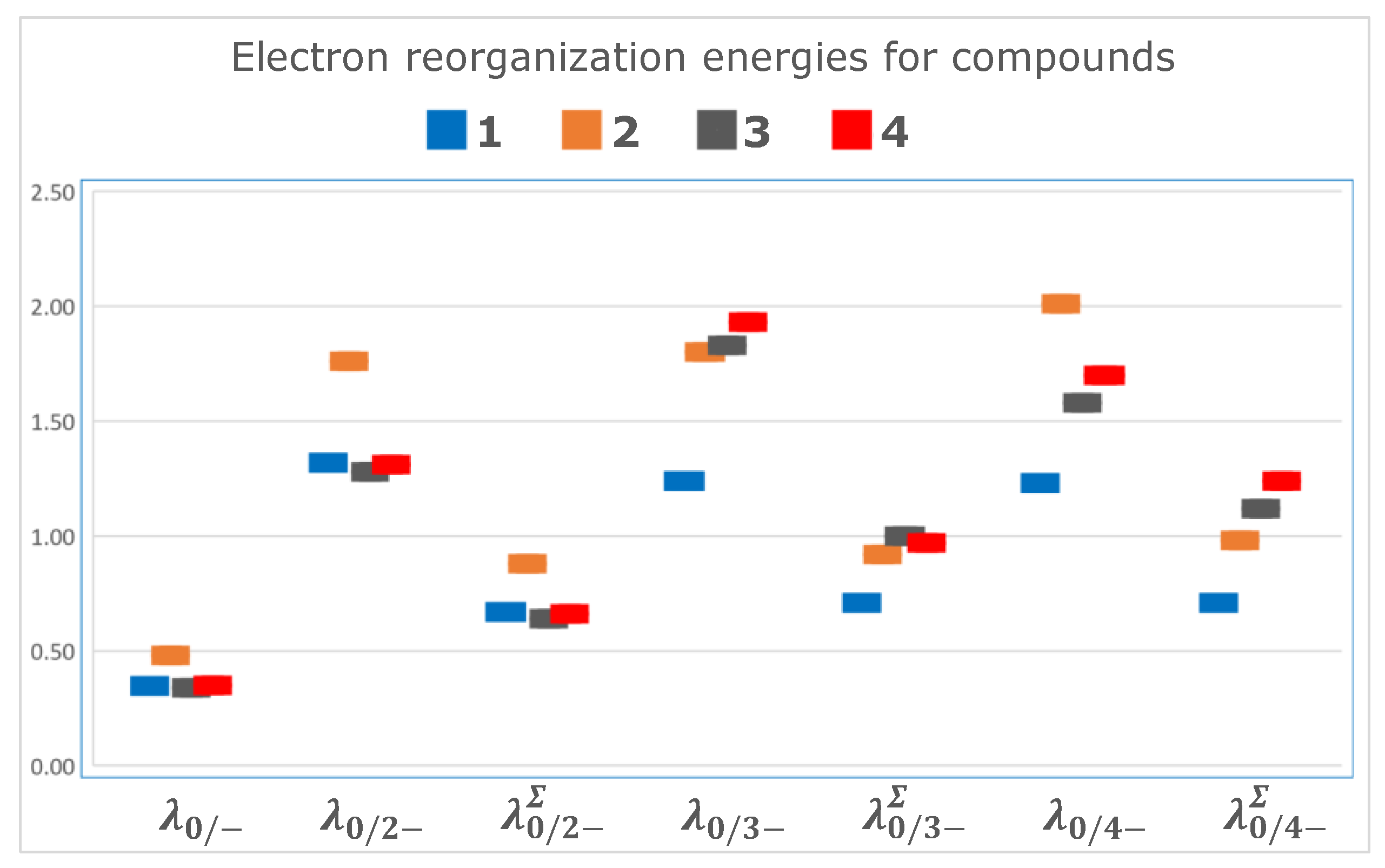
Unlike the values of the reduction potentials, the lambda values turned out to be non-additive when calculated for the consecutive additions of the electrons to successively obtain the mono, di, tri, and tetra-anions. Thus, the values for
,
,
for simultaneous transfers are greater than those calculated from the sum of each one electron transfer (
Figure 4,
Table 5):
The Frank–Condon principle, as applied in the framework of the Marcus theory for charge transfer, provides another via to the evaluation of reorganization energies [
13]. Therefore, for an anion of valence
:
Thus, in case of
, reorganization energy for simultaneous two- and three-electron transfer would be
and
, respectively (
Table 6). These values differ from the ones calculated previously and, therefore, are considered as approximations far from real values.
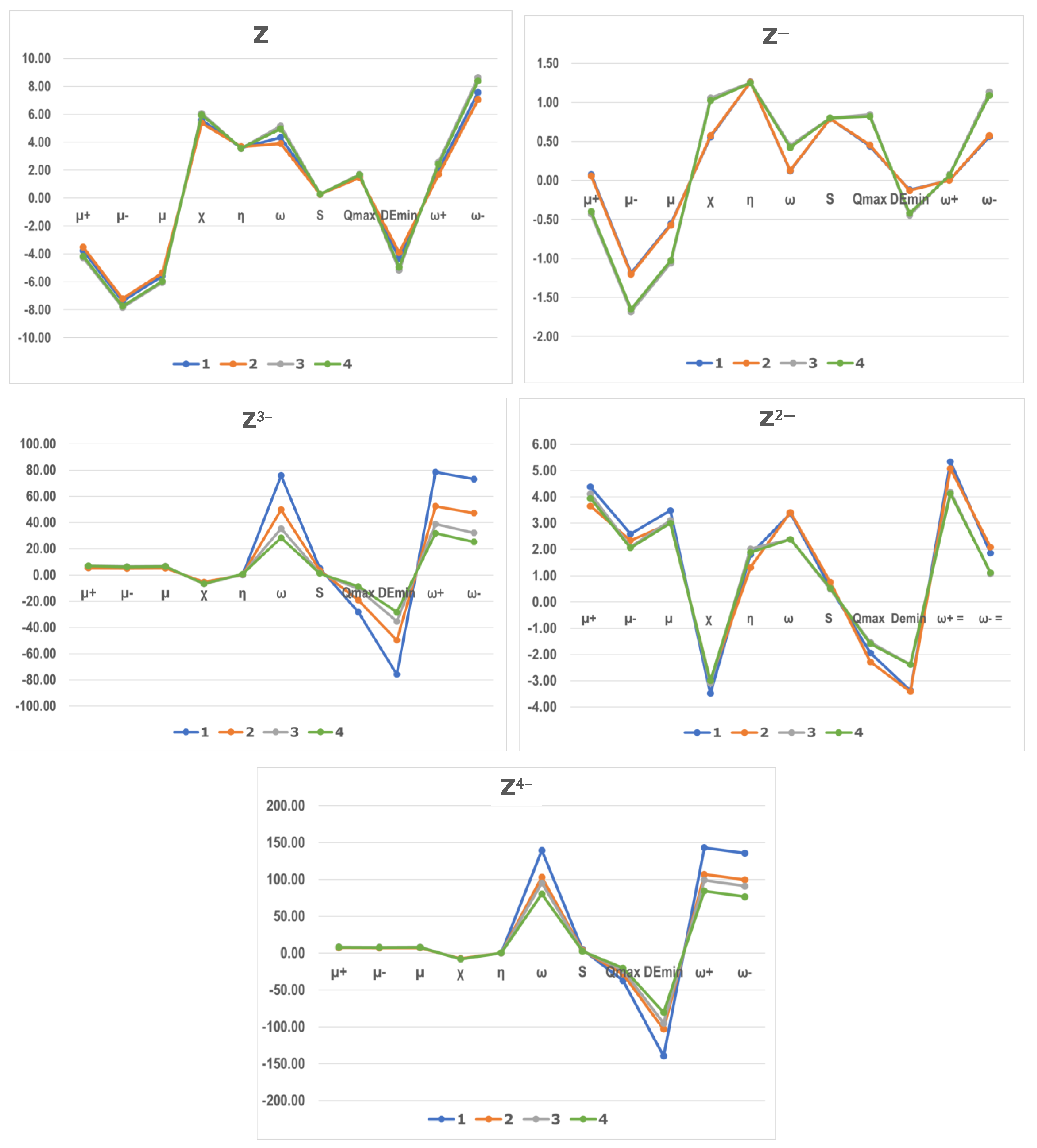
Gabedit 2.5.1 software allowed to determine the Fukui indices. These were calculated using the frontier orbitals of the
Z compounds as neutral, mono-, di-, tri-, and tetra-anions (
Table 7). These values are represented in
Figure 5 to facilitate the comparison of the four compounds studied for each type of anion.
The values of the descriptors of the Fukui indices for the neutral compounds (Z) show only small differences. The differences increase for the mono anionic compounds, establishing similarity on the one hand between 1− and 2− and on the other between 3− and 4−. The dianions Z2− show values inside the order of magnitude of Z and Z−. In the case of Z3− and Z4− the indices: ω (Electrophilicity index), ω−(propensity to donate electron), ω+ (propensity to accept electron), Qmax (Maximal electronic charge accepted by an electrophile), Demin (Energy decrease if the electrophile take Qmax) show values of a larger order of magnitude that led us to think that it is due to a defect in the concept, rather than a trustable prediction.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
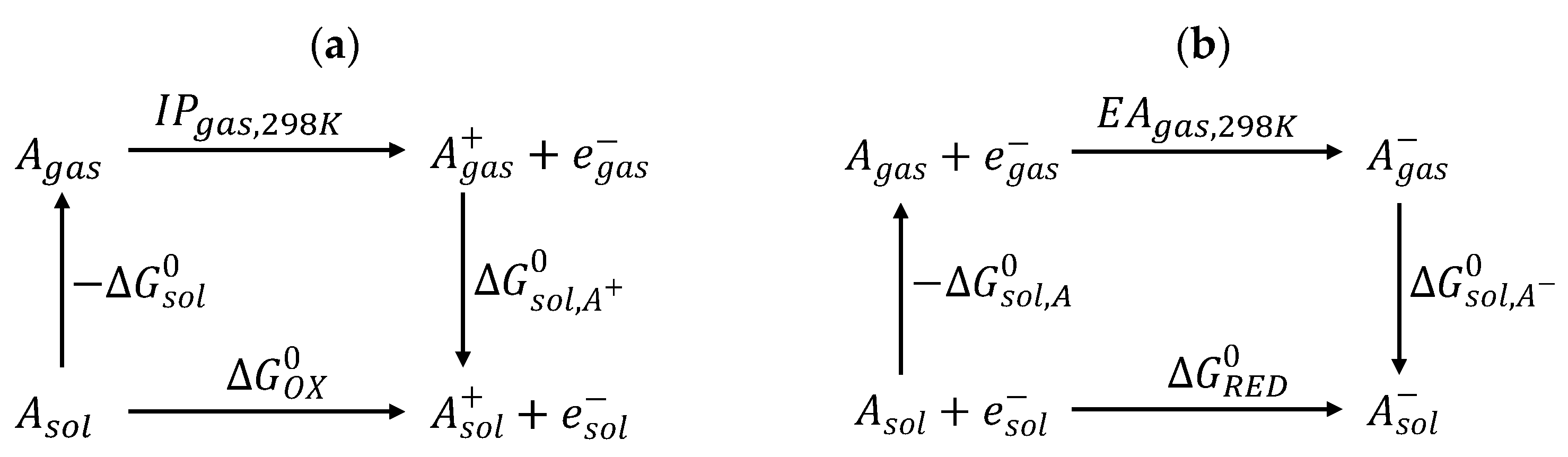{kind=link}
{kind=link}




















 0.1
0.1