1. Introduction
Tuberculosis (TB) is a contagious/infectious disease transmitted through the air that is caused by the fatal pathogen/bacillus Mycobacterium tuberculosis (Mtb). It has been afflicting humans for years and is now causing a global health crisis [
1]. According to a report released by the World Health Organization (WHO) in 2020, tuberculosis caused around 1.4 million fatalities and over 10 million people were ill with the disease in 2019, signifying a devastating influence on global mortality and morbidity rates [
2]. As of today, the WHO believes that one out of every four people has a confirmed tuberculosis infection. MDR TB is a growing pandemic, and the emergence of extended drug-resistant tuberculosis (XDR tuberculosis) offers a new global hazard because it is likely incurable with current medicines. The United Nations Sustainable Development Goals (SDGs) include a health goal of ending the tuberculosis epidemic by 2030 [
3]. To tackle the TB pandemic, innovative treatment medicines with novel modes of action are urgently needed.
The goal can be achieved by employing “conditionally essential target” (CET)-based drug design. M. tuberculosis needs iron to colonise and proliferate as well as to maintain its metabolic machinery [
4]. In human serum and bodily fluids, free iron is severely restricted/significantly low (about 10
−24 M). When faced with a lack of iron in the host, mycobacteria re-uptake tiny molecules known as mycobactins (mycobacterial siderophores/iron chelators) to chelate and ingest this critical trace element from host iron-binding proteins [
5]. The mycobactin production pathway has long been regarded as a promising source of anti-tubercular targets for the creation of new probes. Mycobactin T is highly lipophilic and stays attached to mycobacterial cell walls, whereas carboxymycobactin, which is more polar due to its short carboxylic acid side chain, is released into the extracellular media by the pathogen to chelate the valuable iron element. The main structures of mycobactins are made up of a 2-hydroxyphenyloxazolidine moiety coupled to an acylated -N-hydroxylysine residue esterified with a 3-hydroxybutyric acid at the -carboxyl. The latter forms an amide bond with a second -N-hydroxylysine, which is then cyclised to provide a (seven-membered) lactam [
6]. Following the annotation of the M. tuberculosis genome sequence [
7], a ten-gene locus (mbtA-J) encoding a non-ribosomal peptide synthetase–polyketide synthase (NRPS–PKS) system was shown to be important for the synthesis of the mycobactin peptide core. MbtA, an aryl-adenylating enzyme, catalyses the first two steps of mycobactin biosynthesis. MbtA activates salicylic acid (adenylation step) by producing Sal-AMP, which is subsequently loaded (via an acylation step) onto MtbB’s phosphopantetheinylation domain, which is also part of the NRPS–PKS cluster [
8]. MbtA has no human homologues and has been chemically verified as a target for the development of novel anti-TB agents [
9]. M. tuberculosis mutants that are unable to manufacture mycobactins or import these siderophores with chelated iron have been demonstrated to have reduced virulence and proliferation in the lungs and macrophages in vitro and in vivo. Furthermore, nucleoside antibiotics that selectively block MbtA enzymatic activity, such as 5′-O-[N-(salicyl)sulfamoyl)adenosine (Sal-AMS), were found to successfully stop M. tuberculosis growth and pathogenicity [
10]. Taken together, our findings suggest that drugs targeting the mycobactin production pathway would be effective in the treatment of tuberculosis [
11].
Stirrett et al. [
12] generated a library of small compounds with structural similarities to the mycobactin framework and assessed them against Mtb in 2008, putting the concept of (CET)-based drug design into effect. The 3,5-diaryl-1-carbothioamide-pyrazoline motif, which resembled the hydroxyphenyl-oxazoline unit of the mycobactin and carboxymycobactin siderophores, served as their basic structural scaffold. The compounds were tested for their capacity to suppress Mtb growth in both iron-deficient and iron-rich environments, as well as their ability to block a salicylation enzyme targeted by Sal-AMS. In iron-depleted circumstances, the bactericidal compound (1) had the highest antitubercular activity in the series, with IC
50 and MIC
90 values of 8 and 21 M, respectively. Analog 1 had no cytotoxicity against HeLa cells (CD
50 = 398 M), and it was inactive against Mtb (MIC
90 = 333 M) in iron-rich conditions (GASTD + Fe), showing its involvement in mycobactin system functions, with a remarkable selectivity index (SI
Mtb = IC
50GASTD/IC
50GAST) value of 15. Analog 1’s structure proved to be a suitable platform for lead design against CETs as well as research into the mycobactin synthesis pathway.
![Chemproc 08 00062 i001]()
Ferreras et al. [
13] created a library of mycobactin analogues, including diaryl-substituted pyrazoline (DAP), in 2011 as a follow-up to their previous work. Mtb and Yersinia pestis were tested using DAP derivatives. Compounds 2 and 3 were the most effective against Mtb (IC
50 = 47 M, MIC = 16 M) among the active DAP derivatives. However, the majority of active compounds had anti-tubercular activity in both iron-depleted (GAST) and iron-replete (GAST + Fe) media, implying that the compounds’ targets are linked to mycobacterial activities that are important in both low- and high-iron environments.
![Chemproc 08 00062 i002]()
In line with these previous studies, the goal of this work was to identify novel anti-tubercular drugs with a high affinity for MbtA, the adenylating enzyme that catalyses the first step in mycobactin production and which is only expressed by mycobacteria. In this regard, we attempted to design a library of 12 compounds by altering the pyrazoline moiety (depicted in red) of Ferreras et al.’s reported potent molecules (2 and 3) and modifying the R and R
1 positions, respectively. The designed molecules along with their structures are presented in
Table 1.
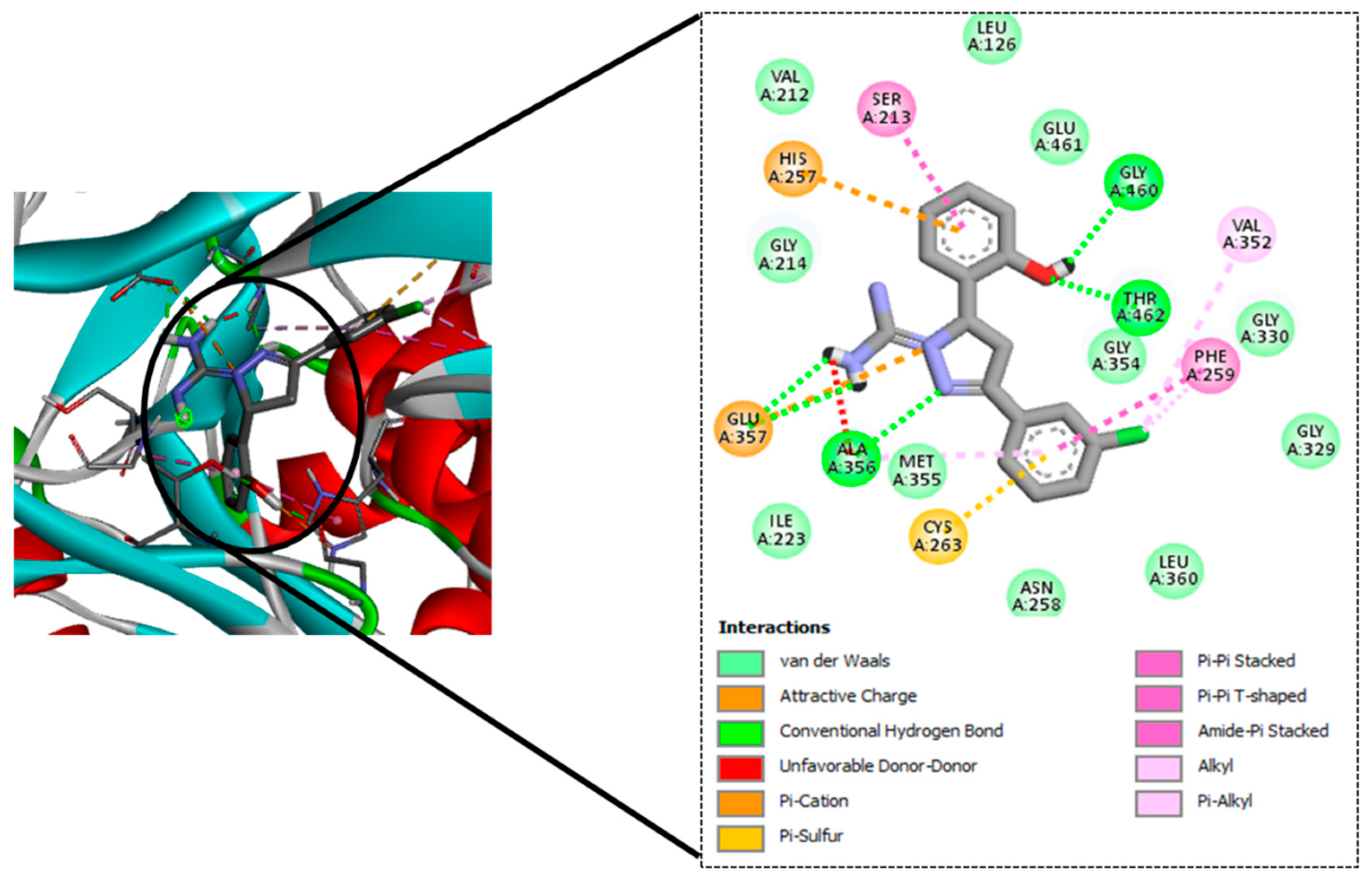
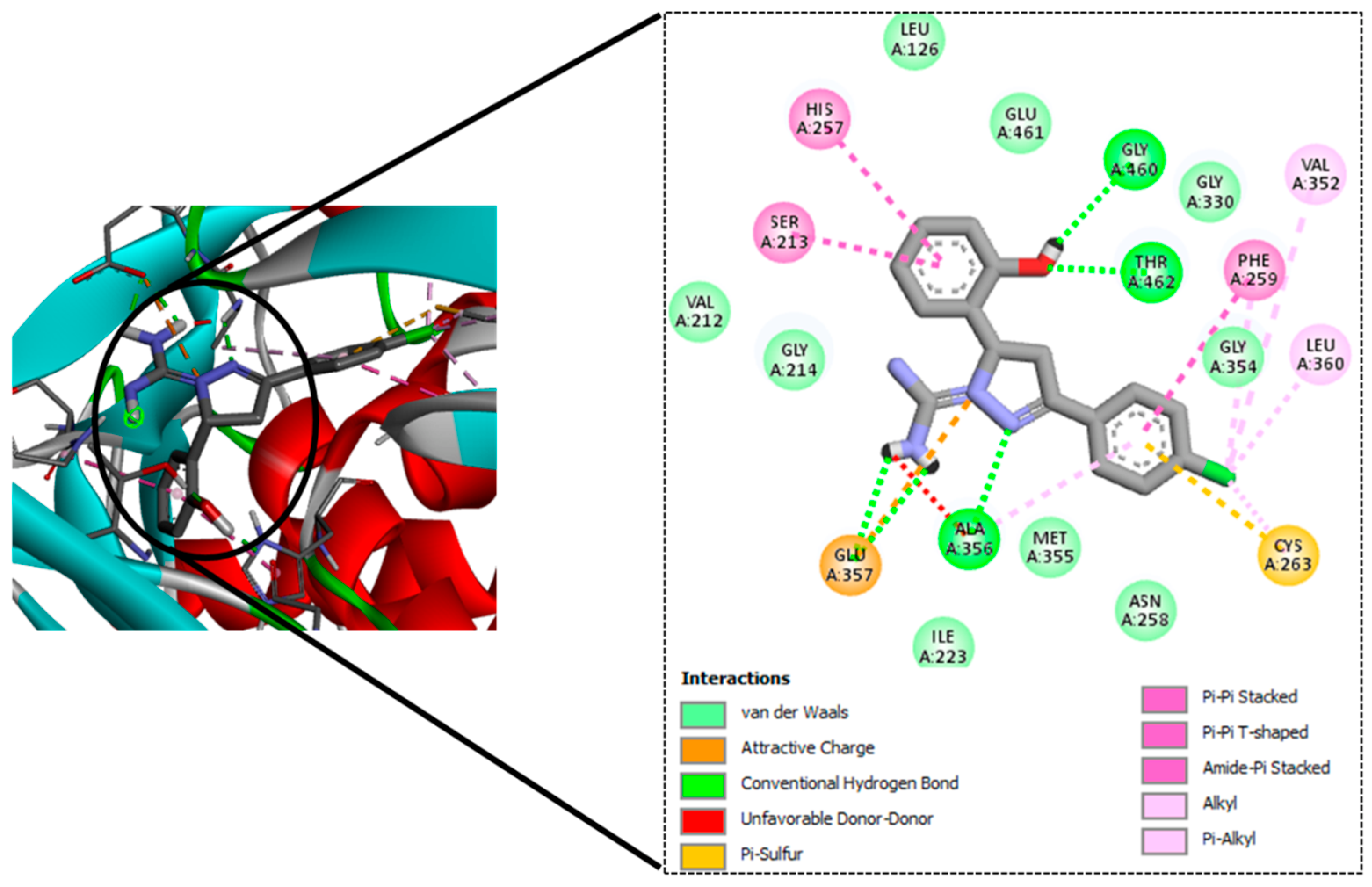
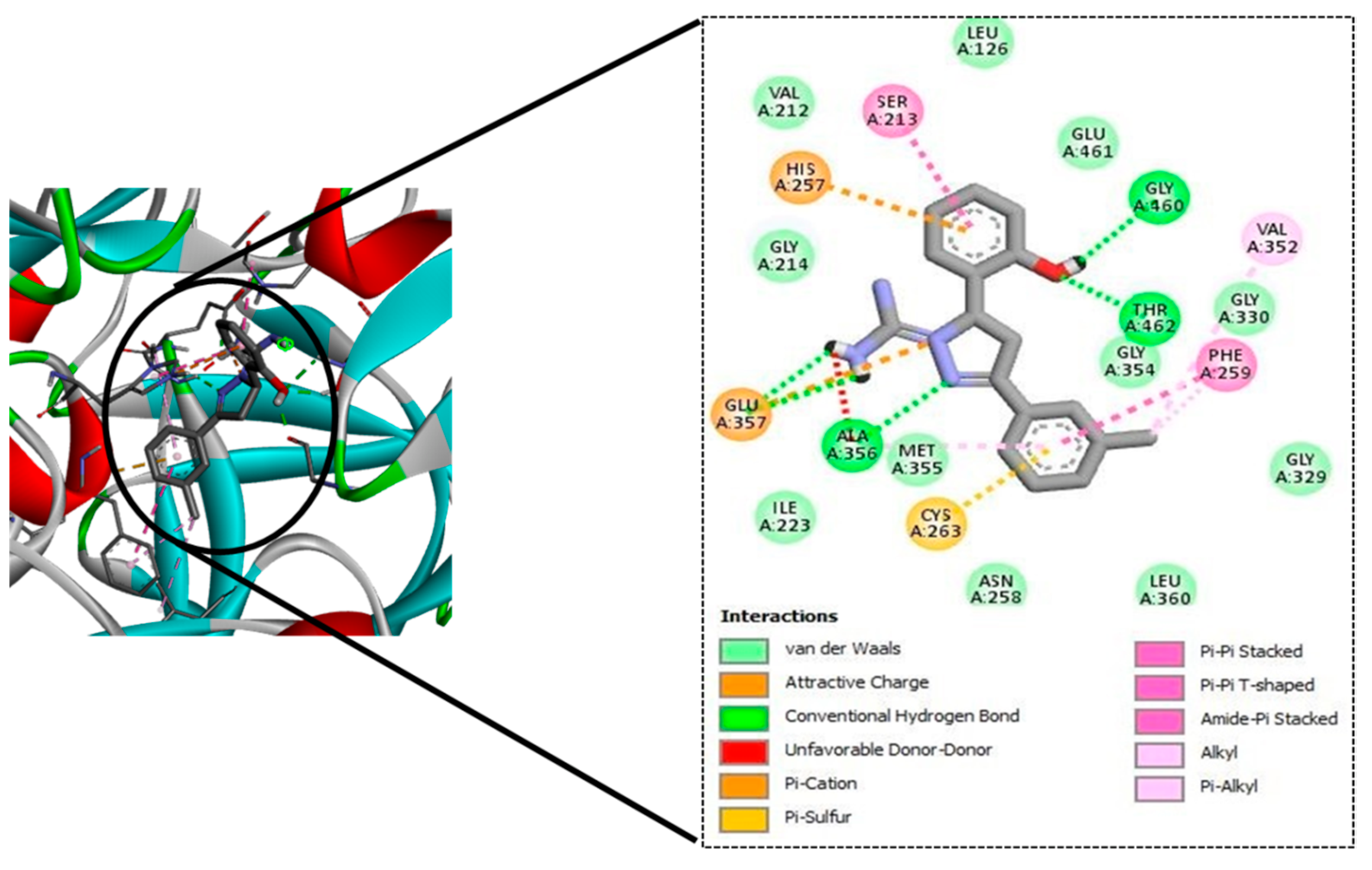
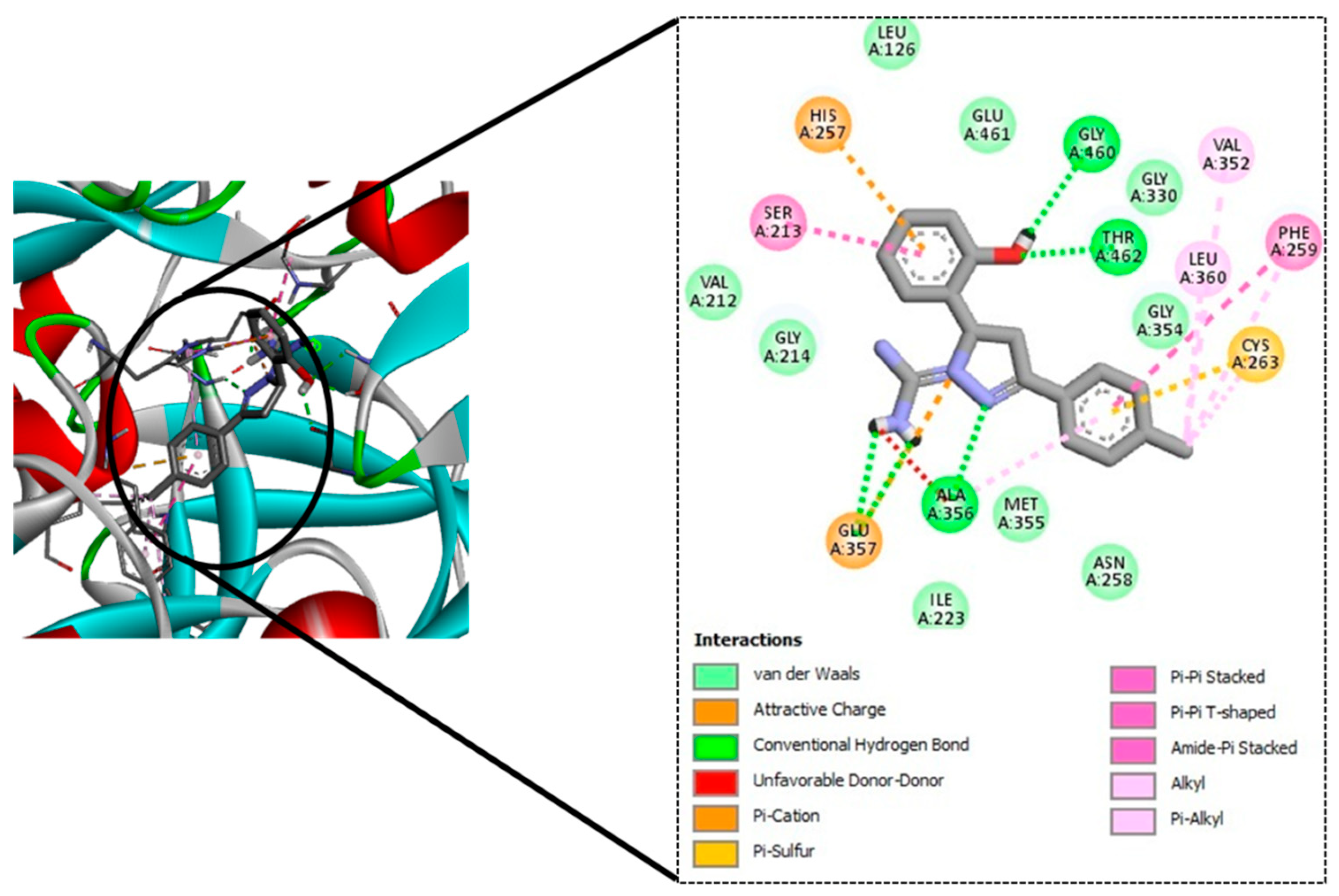
To this end, a 12-member library of designed molecules was tested in silico to test the ability to bind to MbtA. These molecules were docked into the active site of the MbtA crystal structure. The top-scoring molecules were taken up for ADME and toxicity studies.
4. Conclusions
TB remains a substantial health burden in underdeveloped nations, despite significant progress in clinical drug candidate development for TB treatment during the last 10–15 years. The search for therapeutic candidates that inhibit novel targets is still a hot topic in science. Studies aimed at gaining better knowledge of Mtb biology have yielded results, including the discovery of new therapeutic targets. It has been established that mycobacterial virulence and survival in the host are directly affected by impairments in mycobactin production and iron uptake. The rational design of MbtI and MbtA inhibitors based on structure has so far yielded encouraging results. In pursuance of this goal, we used CET-based drug design to find M. tuberculosis inhibitors that can bind to a well-defined target, namely, MbtA. The tubercular enzyme MbtA, a newly discovered TB target that catalyses the initial two-step reaction of mycobactin production, was found to be highly interactive with our top four designed compounds (GM08, GM09, GM02, and GM03). They also showed acceptable pharmacokinetic profiles and nominal toxicity profiles. Furthermore, based on docking scores and predicted pharmacokinetic profiles, it could be concluded that GM08 and GM09 could serve as good leads for future optimization. Exploring intrinsic interactions between potential drugs and their potential therapeutics could open the way for unique and modern antibiotic discovery methodologies to be developed and implemented.











{kind=link}
{kind=link}
{kind=link}
{kind=link}












