
Reaction of N-(tosylmethyl)ureas with NaCN: Synthetic and Mechanistic Aspects †

Abstract
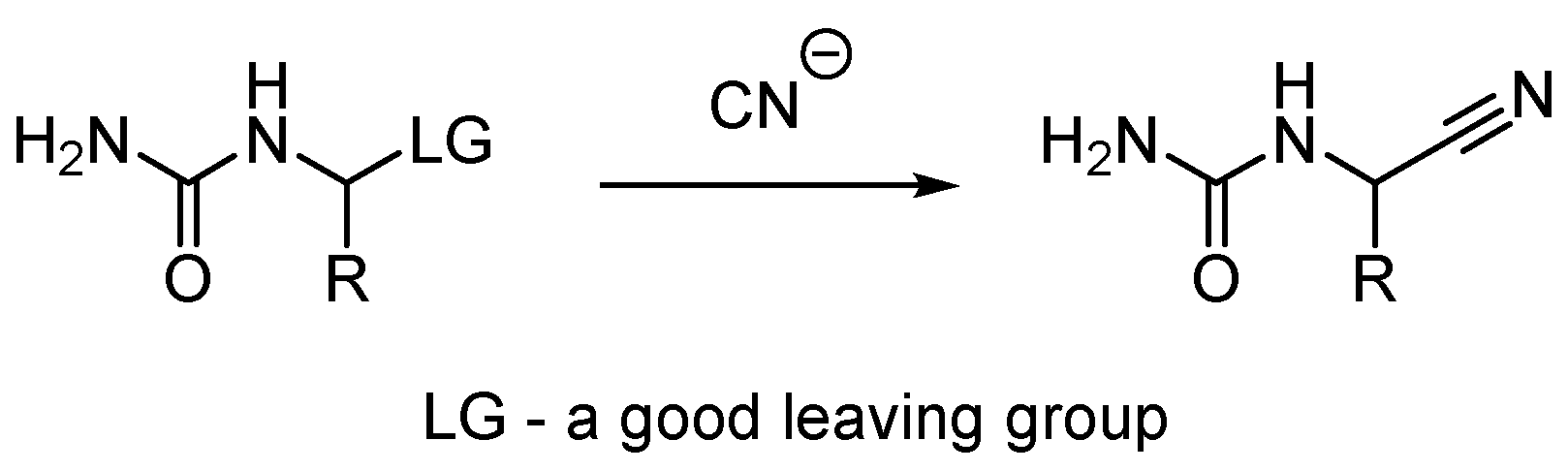
:1. Introduction
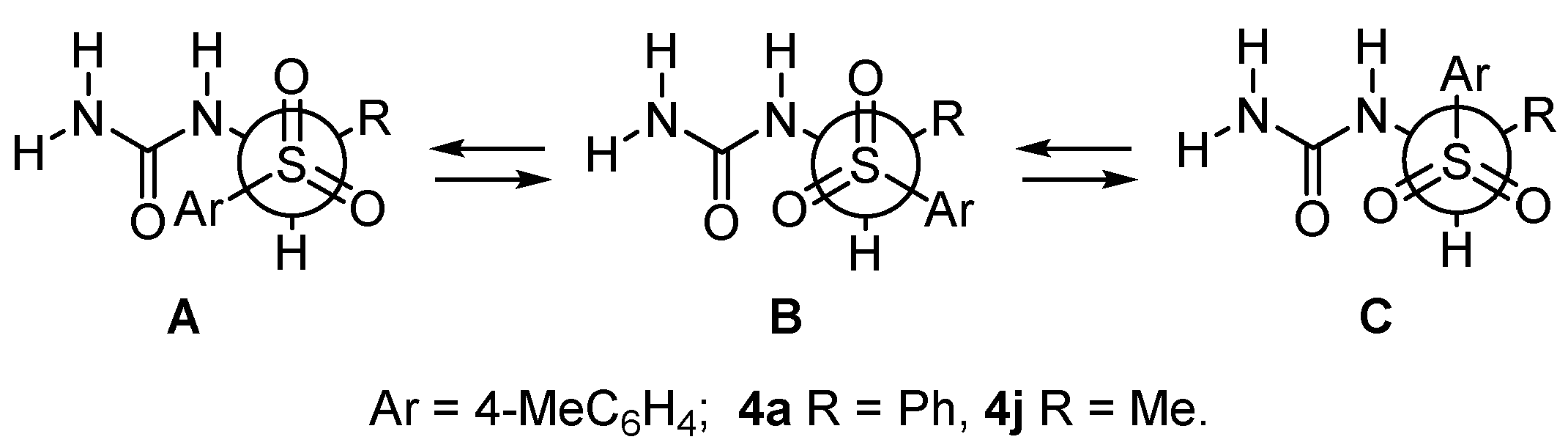
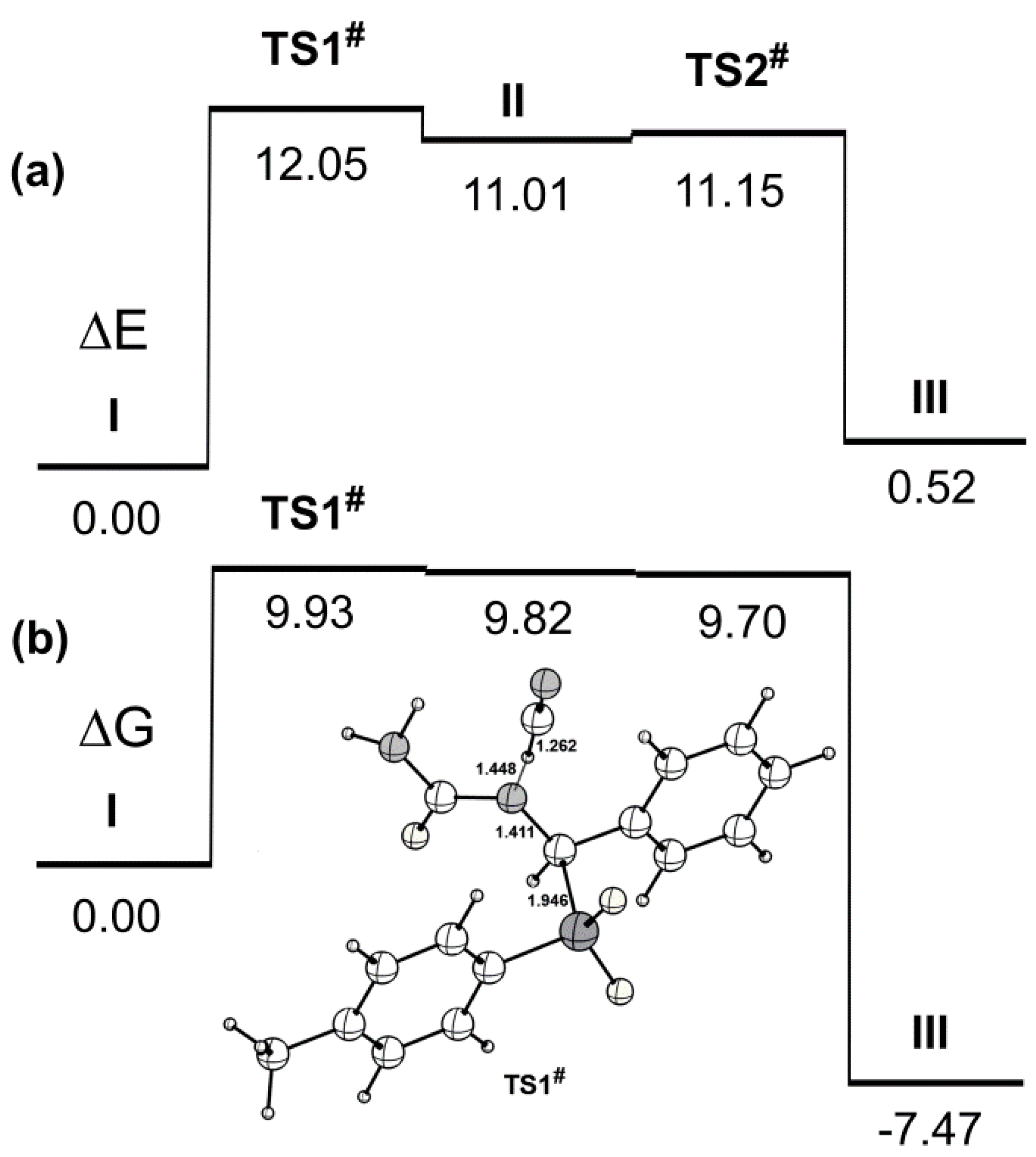
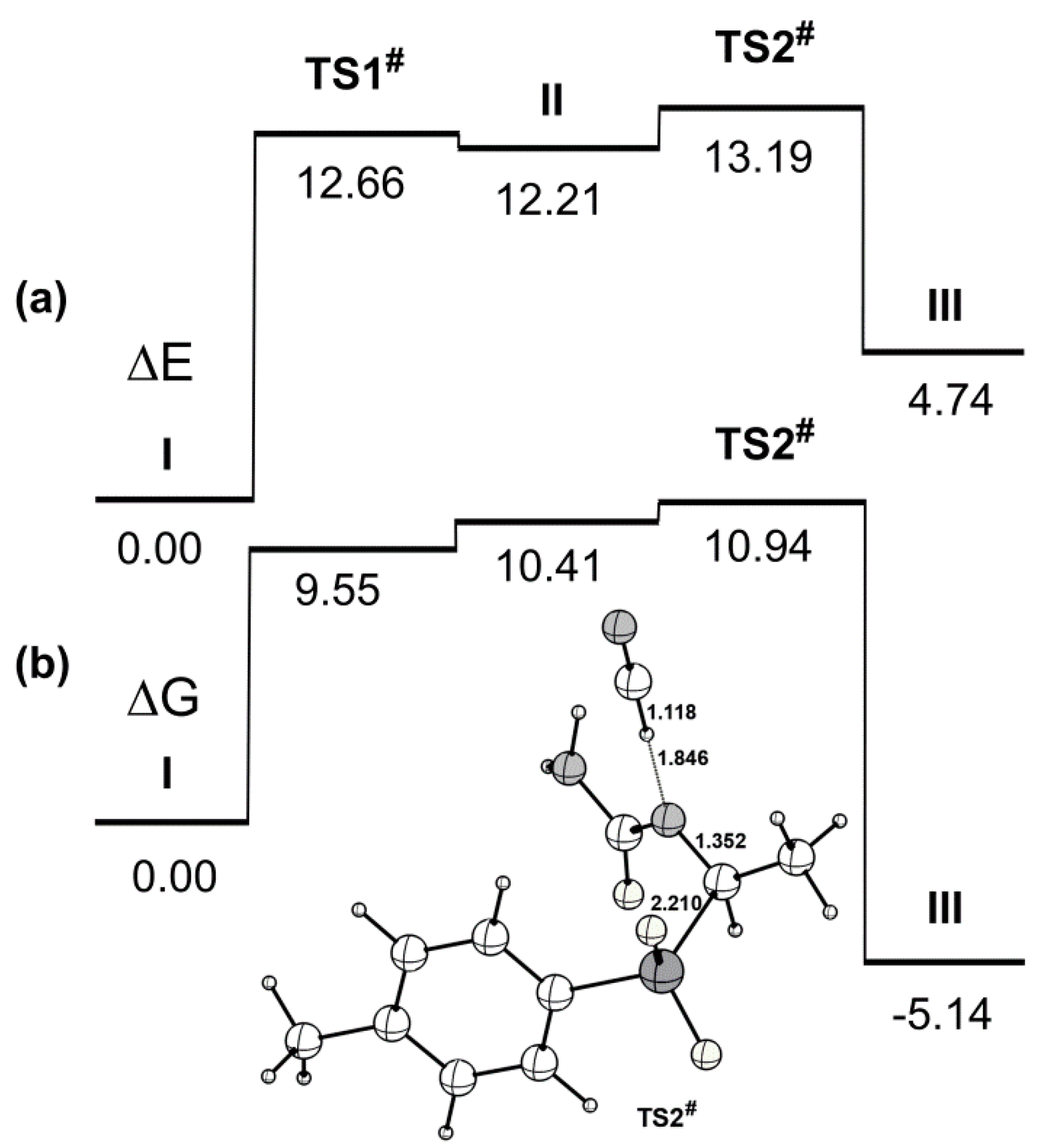
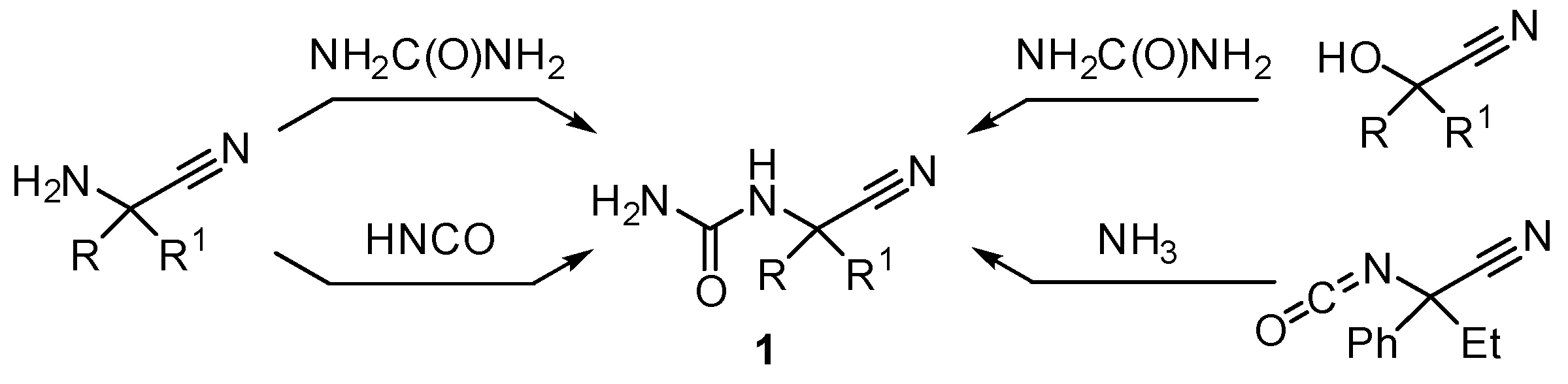
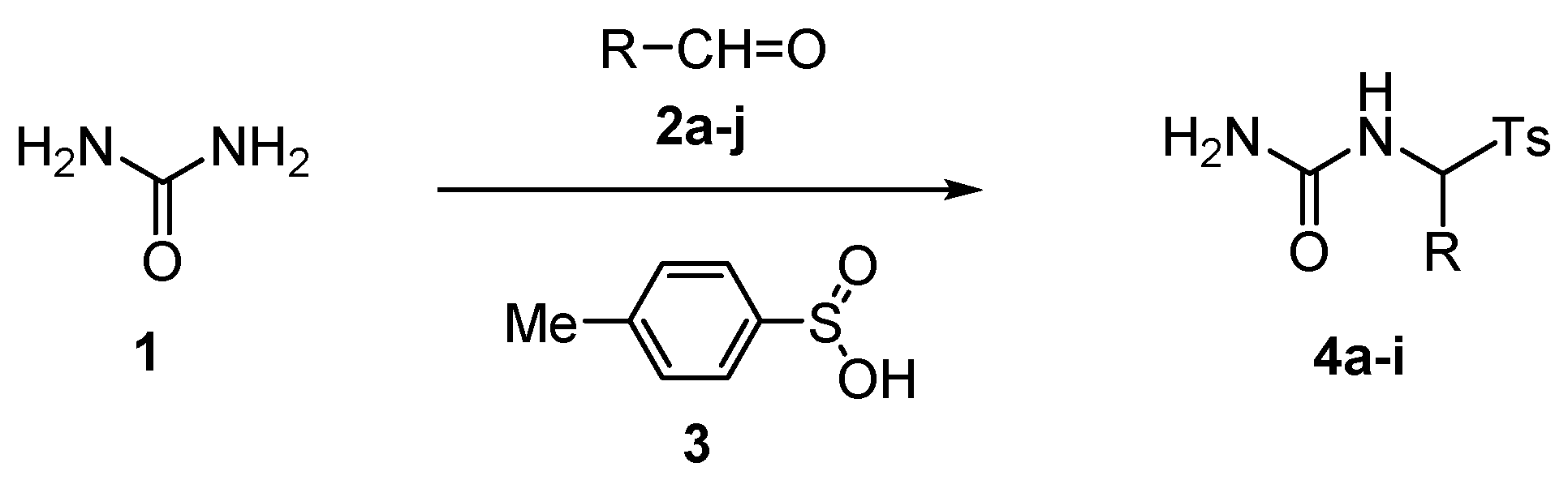
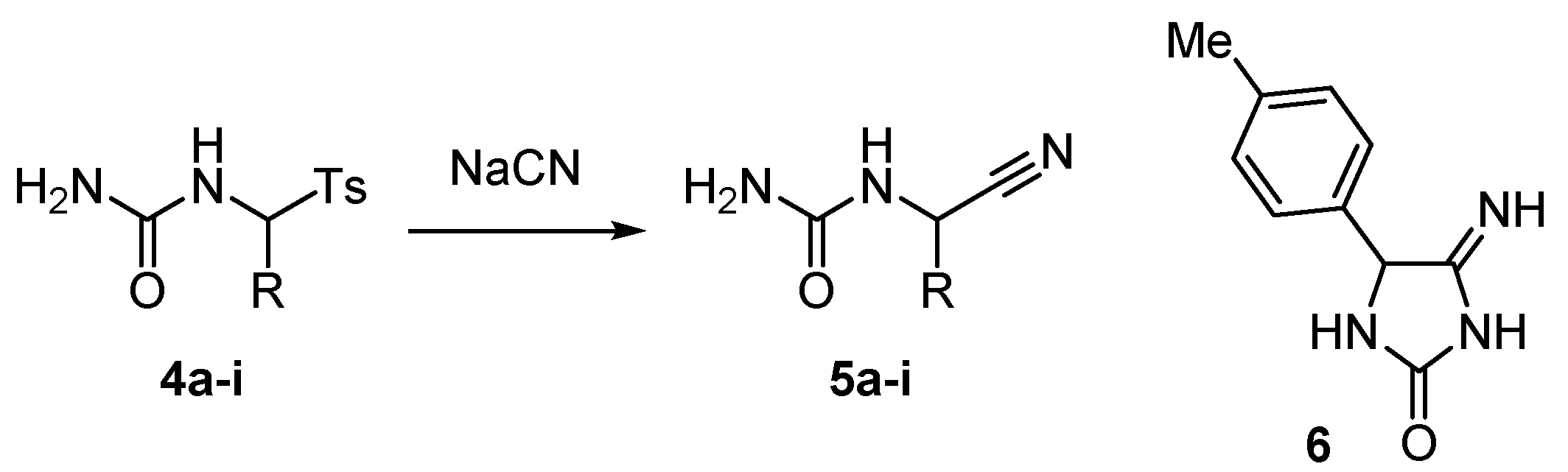
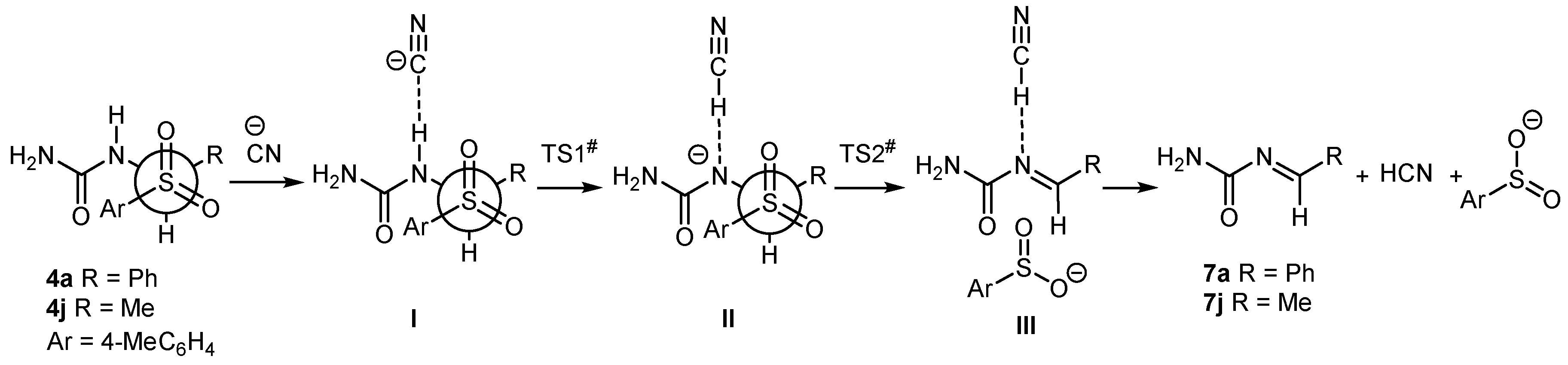
2. Results and Discussion
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schulze, W.; Klepel, M.; Jumar, A.; Lehmann, H.; Braemer, B. N-cyanoalkylurea derivatives as fungicides. Chem. Abstr. 1987, 106, 4685. [Google Scholar]
- Ohishi, T.; Kato, T.; Ozaki, T.; Kameda, N.; Itooka, E.; Fujinami, A. Cyanourea fungicides. Chem. Abstr. 1975, 83, 142996. [Google Scholar]
- Nieper, H.A.; Koehler, F. Cytostatic α-cyanobenzyl derivatives. Chem. Abstr. 1984, 100, 96704. [Google Scholar]
- Schwing, G.W.; Price, W.A., Jr.; Smith, D.H., Jr. Ureidonitrile, Verfahren zu ihrer Herstellung und sie enthaltende Arzneimittel. Chem. Abstr. 1980, 92, 22091. [Google Scholar]
- Löser, R.; Schilling, K.; Dimmig, E.; Guetschow, M. Interaction of Papain-like Cysteine Proteases with Dipeptide-Derived Nitriles. J. Med. Chem. 2005, 48, 7688–7707. [Google Scholar] [CrossRef]
- Salvati, M.E.; Finlay, H.; Harikrishnan, L.S.; Jiang, J.; Johnson, J.A.; Kamau, M.G.; Lawrence, R.M.; Miller, M.M.; Qiao, J.X.; Wang, T.C.; et al. Heterocyclic CETP inhibitors. Chem. Abstr. 2011, 154, 259401. [Google Scholar]
- Giuliano, C.; Rubio, S.G.; Daina, A.; Guainazzi, A.; Pietra, C. p-Substituted Asymmetric Ureas and Medical Uses Thereof. Chem. Abstr. 2015, 163, 412148. [Google Scholar]
- Takahashi, H.; Watanabe, H.; Fujii, K.; Shibasaki, M.; Kawashima, M.; Kamiya, M.; Ohata, K. Urea derivative or pharmacologically acceptable salt thereof. Chem. Abstr. 2016, 166, 39993. [Google Scholar]
- Luengo, J.; Vaddi, K. Selective inhibitors of protein arginine methyltransferase 5 (PRMT5). Chem. Abstr. 2017, 168, 100879. [Google Scholar]
- Moore, J.E. 1-Substituted-3-polyhaloalkylthio hydantoin fungicides. Chem. Abstr. 1987, 106, 98089. [Google Scholar]
- Knabe, J.; Wunn, W. Racem. und optisch aktive Hydantoine aus disubstituierten Cyanessigsäuren. Arch. Pharm. 1980, 313, 538–543. [Google Scholar] [CrossRef]
- Herbst, R.M.; Johnson, T.B. Researches on hydantoins. xlix. a new rearrangement leading to the formation of 4-aminohydantoin derivatives. J. Am. Chem. Soc. 1930, 52, 3676–3680. [Google Scholar] [CrossRef]
- Mas-Roselló, J.; Okoh, M.; Clayden, J. Enantioselectively functionalised phenytoin derivatives by auxiliary-directed N to C aryl migration in lithiated α-amino nitriles. Chem. Commun. 2018, 54, 10985–10988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, J.; Amrein, K.; Hunziker, D.; Kuhn, B.; Mayweg, A.V.; Neidhart, W.; Takahashi, T. Imidazolone and imidazolidinone derivatives as 11B-HSD1 inhibitors for the treatment of diabetes. Chem. Abstr. 2008, 148, 495950. [Google Scholar]
- Cook, A.H.; Hunter, G.D. Studies in the azole series. Part XXXIII. The interaction of α-amino-nitriles and alkyl or aryl isocyanates. J. Chem. Soc. 1952, 3789–3796. [Google Scholar] [CrossRef]
- Husbands, S.; Fraser, W.; Suckling, C.J.; Wood, H.C. Latent inhibitors part 11. The synthesis of 5-spirocyclopropyl dihydroorotic acid. Tetrahedron 1995, 51, 865–870. [Google Scholar] [CrossRef]
- De Simone, A.; Georgiou, C.; Ioannidis, H.; Gupta, A.A.; Juárez-Jiménez, J.; Doughty-Shenton, D.; Blackburn, E.A.; Wear, M.A.; Richards, J.P.; Barlow, P.N.; et al. A computationally designed binding mode flip leads to a novel class of potent tri-vector cyclophilin inhibitors. Chem. Sci. 2019, 10, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Yun, W. Piperidine analogs as glycogen synthase activators. Chem. Abstr. 2011, 154, 588717. [Google Scholar]
- Luo, G.; Chen, L.; Degnan, A.P.; Dubowchik, G.M.; Macor, J.E.; Tora, G.O.; Chaturvedula, P.V. Heterocyclic anti-migraine agents. Chem. Abstr. 2005, 143, 78091. [Google Scholar]
- Coussanes, G.; Gaus, K.; O’Sullivan, A.C. The Synthesis of Ketone-Derived Enamides by Elimination of HCN from Cyanoamides. Eur. J. Org. Chem. 2016, 2016, 4176–4188. [Google Scholar] [CrossRef]
- Labrie, F.; Singh, S.M.; Labrecque, R.; Ciobanu, L.C. Non-steroidal antiandrogens and selective androgen receptor modulators with a pyridyl moiety. Chem. Abstr. 2015, 163, 132721. [Google Scholar]
- Ventosa-Andrés, P.; Vera, J.A.G.; García-López, M.T.; Herranz, R. Solvent-Free Synthesis of α-Amino Nitrile-Derived Ureas. Org. Lett. 2013, 15, 632–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alker, D.; Campbell, S.F.; Cross, P.E.; Burges, R.A.; Carter, A.J.; Gardiner, D.G. Long-acting dihydropyridine calcium antagonists. 5. Synthesis and structure-activity relationships for a series of 2-[[(N-substituted-heterocyclyl)ethoxy]methyl]-1,4-dihydropyridine calcium antagonists. J. Med. Chem. 1990, 33, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Vitali, R.A.; Jacob, T.A.; Chemerda, J.M. Analogs of Methyldopa and Dopa. Hydantoic Acids and Hydantoins. J. Med. Chem. 1964, 7, 379–381. [Google Scholar] [CrossRef]
- Smith, L.H.; Yates, P. The Synthesis of 4-Amino-2(3H)-oxo-5-imidazolecarboxamide. J. Am. Chem. Soc. 1954, 76, 6080–6084. [Google Scholar] [CrossRef]
- Rigler, H.E.; Henze, H.R. Synthesis of Compounds with Hypnotic Properties. I. Alkoxymethylhydantoins. J. Am. Chem. Soc. 1936, 58, 474–477. [Google Scholar] [CrossRef]
- Biltz, H.; Slotta, K. Über die Herstellung von Hydantoinen. J. Prakt. Chem. 1926, 113, 233–267. [Google Scholar] [CrossRef]
- Read, W.T. Researches on hydantoins. synthesis of the soporific, 4,4-phenylethyl-hydantoin(nirvanol). J. Am. Chem. Soc. 1922, 44, 1746–1755. [Google Scholar] [CrossRef] [Green Version]
- Wendelin, W.; Schramm, H.-W. Über die Reaktionen von Guanidin bzw. Harnstoff mit Cyanhydrinen. Chem. Mon. 1981, 112, 853–866. [Google Scholar] [CrossRef]
- Pinner, A.; Lifschütz, J. Ueber die Einwirkung von Harnstoff auf Cyanhydrine. Eur. J. Inorg. Chem. 1887, 20, 2351–2358. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, H. Schiff base metal manganese complex and preparation method and application thereof. Chem. Abstr. 2016, 165, 283533. [Google Scholar]
- Yan, H.; Oh, J.S.; Lee, J.; Song, C.E. Scalable organocatalytic asymmetric Strecker reactions catalysed by a chiral cyanide generator. Nat. Commun. 2012, 3, 1212. [Google Scholar] [CrossRef] [Green Version]
- Reingruber, R.; Baumann, T.; Dahmen, S.; Bräse, S. Use of the Chiral Pool—Practical Asymmetric Organocatalytic Strecker Reaction with Quinine. Adv. Synth. Catal. 2009, 351, 1019–1024. [Google Scholar] [CrossRef]
- Ooi, T.; Uematsu, Y.; Fujimoto, J.; Fukumoto, K.; Maruoka, K. Advantage of in situ generation of N-arylsulfonyl imines from α-amide sulfones in the phase-transfer-catalyzed asymmetric Strecker reaction. Tetrahedron Lett. 2007, 48, 1337–1340. [Google Scholar] [CrossRef]
- Banphavichit, V.; Chaleawlertumpon, S.; Bhanthumnavin, W.; Vilaivan, T. A Convenient Synthesis of N-Boc-Protected α-Aminonitriles from α-Amidosulfones. Synth. Commun. 2004, 34, 3147–3160. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Urogdi, L. Benzotriazole-assisted synthesis of α-acylaminonitriles and a conceptually novel method for peptide elongation. J. Chem. Soc. Perkin Trans. 1 1990, 1, 1853–1857. [Google Scholar] [CrossRef]
- Hu, X.; Li, R.; Li, Z. Conversion of N-Benzyloxycarbonylamino- and N-Tosylamino-Benzyl Phenylsulfones by Green Strecker Reactions to α-Aminobenzyl Nitriles Using Potassium Hexacyanoferrate(II). J. Chem. Res. 2014, 38, 432–436. [Google Scholar] [CrossRef]
- Herrera, R.P.; Sgarzani, V.; Bernardi, L.; Fini, F.; Pettersen, D.; Ricci, A. Phase Transfer Catalyzed Enantioselective Strecker Reactions of α-Amido Sulfones with Cyanohydrins. J. Org. Chem. 2006, 71, 9869–9872. [Google Scholar] [CrossRef]
- Oba, M.; Koguchi, S.; Nishiyama, K. Stereoselective Synthesis of 3,4-Dihydroxylated Prolines and Prolinols Starting fromL-Tartaric Acid. J. Heterocycl. Chem. 2014, 51, 237–243. [Google Scholar] [CrossRef]
- Kadam, S.T.; Thirupathi, P.; Kim, S.S. Synthetic application of in situ generation of N-acyliminium ions from α-amido p-tolylsulfones for the synthesis of α-amino nitriles. Tetrahedron 2010, 66, 1684–1688. [Google Scholar] [CrossRef]
- Das, B.; Damodar, K.; Shashikanth, B.; Srinivas, Y.; Kalavathi, I. A Mild and Efficient Catalytic Strecker Reaction of N-Alkoxycarbonylamino Sulfones with Trimethylsilyl Cyanide Using Indium(III) Chloride: A Facile Synthesis of α-Aminonitriles. Synlett 2008, 2008, 3133–3136. [Google Scholar] [CrossRef]
- Harding, K.E.; Liu, L.T.; Farrar, D.G.; Coleman, M.T.; Tansey, S.K. Preparation of AcyclicN-Acyl-N,O-acetals by Decarboxylation ofN-Protected α-Amino Acids and Studies of Asymmetric Amidoalkylation with Trimethylsilyl Cyanide. Synth. Commun. 1991, 21, 1409–1417. [Google Scholar] [CrossRef]
- Shutalev, A.D. Reaction of α-(thio)amidoalkylation in the synthesis of α-cyano-substituted cyclic (thio)ureas and dithiocarbamates. Chem. Heterocycl. Compd. 1993, 29, 1192–1199. [Google Scholar] [CrossRef]
- Fesenko, A.A.; Grigoriev, M.S.; Shutalev, A.D. Nucleophile-Mediated Ring Expansion of 5-Acyl-substituted 4-Mesyloxymethyl-1,2,3,4-tetrahydropyrimidin-2-ones in the Synthesis of 7-Membered Analogues of Biginelli Compounds and Related Heterocycles. J. Org. Chem. 2017, 82, 8085–8110. [Google Scholar] [CrossRef]
- Fesenko, A.A.; Shutalev, A.D. Synthesis of γ-Azido-β-ureido Ketones and Their Transformation into Functionalized Pyrrolines and Pyrroles via Staudinger/aza-Wittig Reaction. J. Org. Chem. 2013, 78, 1190–1207. [Google Scholar] [CrossRef]
- Shutalev, A.D.; Fesenko, A.A. 4-Hydroxy-4-methyl-5-tosylhexahydropyrimidin-2-imines: Synthesis and different dehydration pathways. Tetrahedron 2011, 67, 6883–6888. [Google Scholar] [CrossRef]
- Fesenko, A.A.; Cheshkov, D.A.; Shutalev, A.D. Synthesis of diethyl 2-thioxo-1,2,3,4-tetrahydro- and hexahydropyrimidine-5-phosphonates. Mendeleev Commun. 2008, 18, 51–53. [Google Scholar] [CrossRef]
- Meijer, H.; Tel, R.M.; Strating, J.; Engberts, J.B.F.N. Mannich-type condensation reactions of sulfinic acids with aldehydes and nitrogen bases: Part. VI. The use of ureas, thioureas and thiobenzamide. Recl. Trav. Chim. 1973, 92, 72–82. [Google Scholar] [CrossRef]
- Zaugg, H.E.; Martin, W.B. α-Amidoalkylations at Carbon. Org. React. 1965, 14, 52–269. [Google Scholar]
- Zaugg, H.E. Recent Synthetic Methods Involving Intermolecular α-Amidoalkylation at Carbon. Synthesis 1970, 1970, 49–73. [Google Scholar] [CrossRef]
- Zaugg, H.E. α-Amidoalkylation at Carbon: Recent Advances—Part I. Synthesis 1984, 1984, 85–110. [Google Scholar] [CrossRef]
- Zaugg, H.E. Synthesis α-Amidoalkylation at Carbon: Recent Advances—Part II. Synthesis 1984, 1984, 181–212. [Google Scholar] [CrossRef]
- Speckamp, W.; Moolenaar, M.J. New Developments in the Chemistry of N-Acyliminium Ions and Related Intermediates. Tetrahedron 2000, 56, 3817–3856. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Zhang, H.-C.; Cohen, J.H.; Turchi, A.I.J.; Maryanoff, C.A. Cyclizations of N-Acyliminium Ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef]
- Petrini, M. α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives. Chem. Rev. 2005, 105, 3949–3977. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Adamek, J.; Zielińska, K. α-Amidoalkylating Agents: Structure, Synthesis, Reactivity and Application. Adv. Heterocycl. Chem. 2014, 111, 43–94. [Google Scholar]
- Wu, P.; Nielsen, T.E. Scaffold Diversity from N-Acyliminium Ions. Chem. Rev. 2017, 117, 7811–7856. [Google Scholar] [CrossRef]
- Marcantoni, E.; Palmieri, A.; Petrini, M. Recent synthetic applications of α-amido sulfones as precursors of N-acylimino derivatives. Org. Chem. Front. 2019, 6, 2142–2182. [Google Scholar] [CrossRef]
- Lillo, V.J.; Mansilla, J.; Saá, J.M. Organocatalysis by Networks of Cooperative Hydrogen Bonds: Enantioselective Direct Mannich Addition to Preformed Arylideneureas. Angew. Chem. Int. Ed. 2016, 55, 4312–4316. [Google Scholar] [CrossRef]
- Lillo, V.J.; Mansilla, J.; Saá, J.M. The role of proton shuttling mechanisms in solvent-free and catalyst-free acetalization reactions of imines. Org. Biomol. Chem. 2018, 16, 4527–4536. [Google Scholar] [CrossRef]
- Davis, T.A.; Vilgelm, A.E.; Richmond, A.; Johnston, J.N. Preparation of (−)-Nutlin-3 Using Enantioselective Organocatalysis at Decagram Scale. J. Org. Chem. 2013, 78, 10605–10616. [Google Scholar] [CrossRef] [Green Version]
- Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. Heterobimetallic Transition Metal/Rare Earth Metal Bifunctional Catalysis: A Cu/Sm/Schiff Base Complex for Syn-Selective Catalytic Asymmetric Nitro-Mannich Reaction. J. Am. Chem. Soc. 2010, 132, 4925–4934. [Google Scholar] [CrossRef]
- Rudolph, F.A.M.; Fuller, A.L.; Slawin, A.M.Z.; Bühl, M.; Aitken, R.A.; Woollins, J.D. The X-ray Structures of Sulfones. J. Chem. Crystallogr. 2009, 40, 253–265. [Google Scholar] [CrossRef]
- Sands, D.E. The crystal structure of dimethyl sulfone. Z. Kristallogr. 1963, 119, 245–251. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
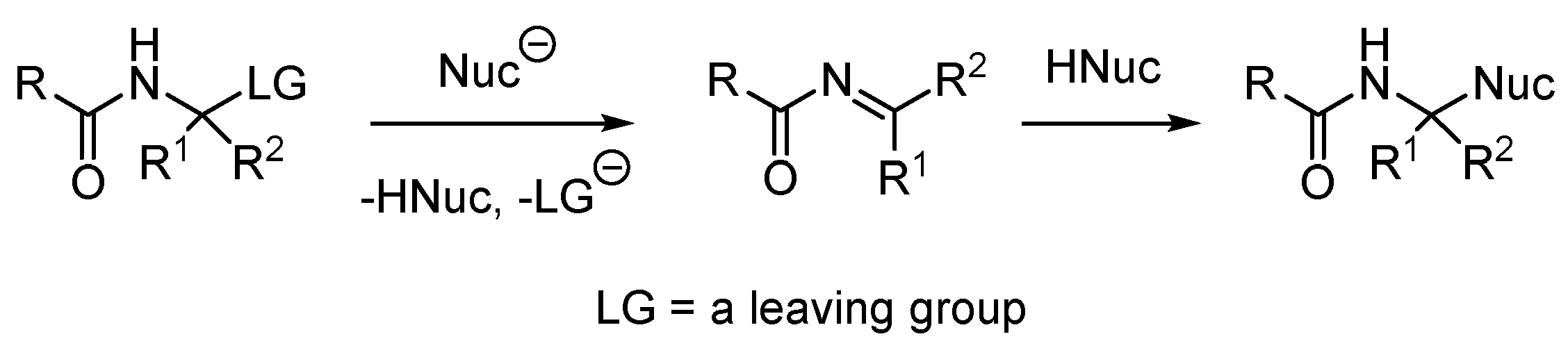{kind=link}
{kind=link}
| Entry | Aldehyde (R) | 1:2:3 Molar Ratio | Solvent | Time (h) | Product | Yield (%) b |
|---|---|---|---|---|---|---|
| 1 | 2a (Ph) | 5:1:1 | H2O | 20 | 4a | 94 |
| 2 | 2a (Ph) | 5:1:1 | 25% aq EtOH | 20 | 4a | 94 |
| 3 | 2b (4-MeC6H4) | 5:1:1 | H2O | 20 | 4b | 97 |
| 4 | 2c (4-EtC6H4) | 5:1:1 | H2O | 20 | 4c | 98 |
| 5 | 2d (4-MeOC6H4) | 5:1:1 | H2O | 20 | 4d | 97 |
| 6 | 2d (4-MeOC6H4) | 5:1:1 | 25% aq EtOH | 24 | 4d | 96 |
| 7 | 2e (3,4-(MeO)2C6H3) | 5:1:1 | H2O | 24 | 4e | 97 |
| 8 | 2e (3,4-(MeO)2C6H3) | 5:1:1 | 25% aq EtOH | 24 | 4e | 98 |
| 9 | 2e (3,4-(MeO)2C6H3) | 5:1:1 | 21% aq HCOOH | 24 | 4e | 99 |
| 10 | 2f (fur-2-yl) | 5:1:1 | H2O | 20 | 4f | 96 |
| 11 | 2g (Pr) | 3:1:1 | H2O | 6.25 | 4g c | 89 |
| 12 | 2g (Pr) | 3:1:1 | H2O | 2 | 4g d | 94 |
| 13 | 2g (Pr) | 5:1:1 | H2O | 2 | 4g | 96 |
| 14 | 2h (i-Pr) | 3:1:1 | H2O | 7.33 | 4h e | 80 |
| 15 | 2h (i-Pr) | 3:1:1 | H2O | 2.4 | 4h f | 88 |
| 16 | 2h (i-Pr) | 5:1:1 | H2O | 2 | 4h | 90 |
| 17 | 2i (Bu) | 5:1:1 | H2O | 2 | 4i | 91 |
| Entry | 4 | R | Equiv. of NaCN | Reaction Conditions | Product | Yield (%) a |
|---|---|---|---|---|---|---|
| 1 | 4a | Ph | 1.10 | DMF, 0 °C, 1 h | 5a | 83 |
| 2 | 4a | Ph | 2.00 | MeCN, rt, 73 h | 5a | 81 |
| 3 b | 4a | Ph | 1.51 | MeCN, rt, 9.75 h | 5a | 73 |
| 4 | 4b | 4-MeC6H4 | 1.25 | DMF, rt, 3 h | 5b c | 85 |
| 5 | 4b | 4-MeC6H4 | 1.22 | DMF, rt, 1 h | 5b d | 91 |
| 6 | 4b | 4-MeC6H4 | 1.21 | DMF, 0 °C, 1 h | 5b e | 92 |
| 7 | 4b | 4-MeC6H4 | 1.10 | DMF, 0 °C, 1 h | 5b | 97 |
| 8 | 4b | 4-MeC6H4 | 2.00 | MeCN, rt, 113 h | 5b | 97 |
| 9 | 4c | 4-EtC6H4 | 1.11 | DMF, 0 °C, 1 h | 5c | 100 |
| 10 | 4d | 4-MeOC6H4 | 1.11 | DMF, 0 °C, 1 h | 5d | 96 |
| 11 | 4e | 3,4-(MeO)2C6H3 | 1.11 | DMF, 0 °C, 1 h | 5e | 95 |
| 12 | 4f | fur-2-yl | 1.10 | DMF, 0 °C, 1 h | 5f | 75 |
| 13 | 4f | fur-2-yl | 1.12 | DMSO, rt, 1 h | 5f | 69 |
| 14 | 4g | Pr | 1.10 | DMF, 0 °C, 1 h | 5g | 69 |
| 15 b | 4g | Pr | 1.52 | MeCN, rt, 8.67 h | 5g | 0 f |
| 16 | 4h | i-Pr | 1.12 | DMF, 0 °C, 1 h | 5h | 0 g |
| 17 | 4i | Bu | 1.10 | DMF, 0 °C, 1 h | 5i | 71 |
| Parameter | 4a | 4j | ||||
|---|---|---|---|---|---|---|
| A | B | C | A | B | C | |
| ΔE, kcal/mol | 0.00 | 0.49 | 0.90 | 0.00 | 0.51 | 0.70 |
| ΔG, kcal/mol | 0.00 | 0.81 | 0.36 | 0.00 | 0.42 | 1.35 |
| C-S, Ǻ | 1.907 | 1.915 | 1.926 | 1.889 | 1.903 | 1.896 |
| C-N, Ǻ | 1.425 | 1.425 | 1.420 | 1.424 | 1.422 | 1.423 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fesenko, A.A.; Shutalev, A.D. Reaction of N-(tosylmethyl)ureas with NaCN: Synthetic and Mechanistic Aspects. Chem. Proc. 2022, 8, 54. https://doi.org/10.3390/ecsoc-25-11759
Fesenko AA, Shutalev AD. Reaction of N-(tosylmethyl)ureas with NaCN: Synthetic and Mechanistic Aspects. Chemistry Proceedings. 2022; 8(1):54. https://doi.org/10.3390/ecsoc-25-11759
Chicago/Turabian StyleFesenko, Anastasia A., and Anatoly D. Shutalev. 2022. "Reaction of N-(tosylmethyl)ureas with NaCN: Synthetic and Mechanistic Aspects" Chemistry Proceedings 8, no. 1: 54. https://doi.org/10.3390/ecsoc-25-11759