
Quantitative Structure–Property Relationship for the Retention Index of Volatile and Semi-Volatile Compounds of Coffee †

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Description
2.2. Molecular Representation and Geometry Optimization
2.3. Molecular Descriptor Calculation and Reduction
2.4. Molecular Descriptor Selection
2.5. Validation of the Model
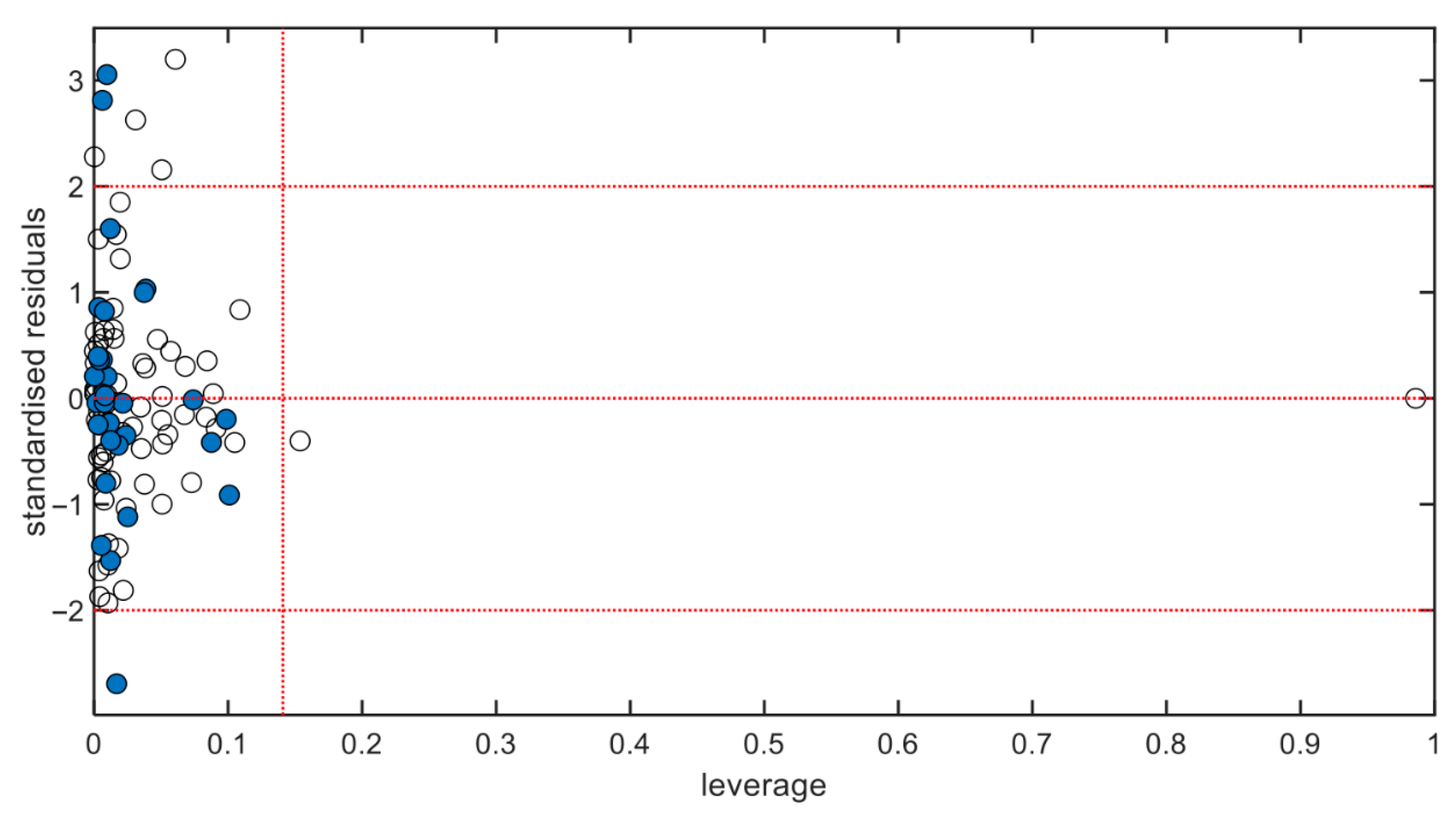
2.6. Applicability Domain
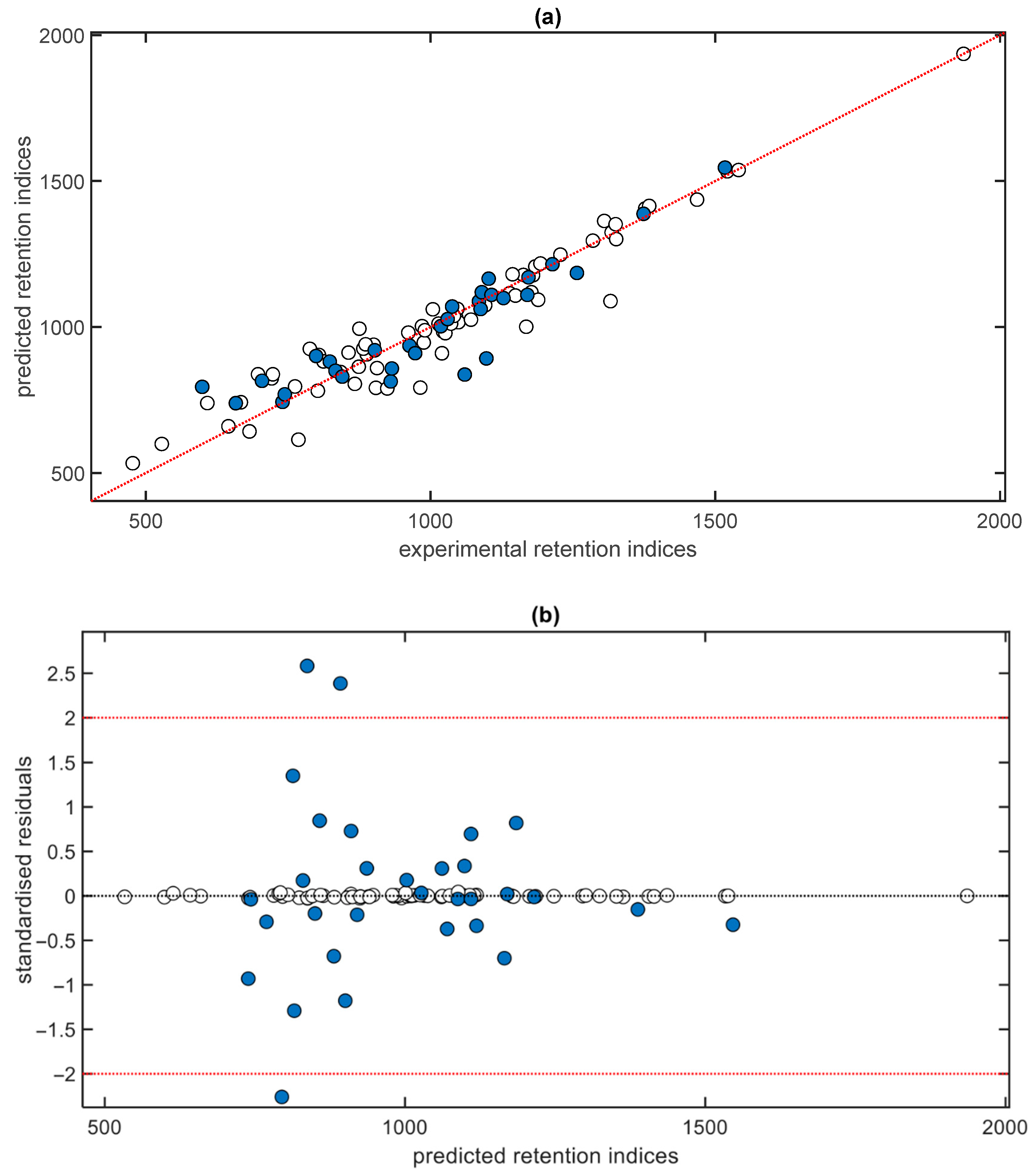
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandes, A.P.; Santos, M.C.; Lemos, S.G.; Ferreira, M.M.; Nogueira, A.R.A.; Nóbrega, J.A. Pattern recognition applied to mineral characterization of Brazilian coffees and sugar-cane spirits. Spectrochim. Acta B: At. Spectrosc. 2005, 60, 717–724. [Google Scholar] [CrossRef]
- Dos Santos, É.J.; de Oliveira, E. Determination of mineral nutrients and toxic elements in Brazilian soluble coffee by ICP-AES. J. Food Compos. Anal. 2001, 14, 523–531. [Google Scholar] [CrossRef]
- Torga, G.N.; Spers, E.E. Perspectives of global coffee demand. In Coffee Consumption and Industry Strategies in Brazil; de Almeida, L.F., Spers, E.E., Eds.; Woodhead Publishing: Duxford, UK, 2020; pp. 21–49. [Google Scholar]
- de Melo Pereira, G.V.; de Carvalho Neto, D.P.; Júnior, A.I.M.; do Prado, F.G.; Pagnoncelli, M.G.B.; Karp, S.G.; Soccol, C.R. Chemical composition and health properties of coffee and coffee by-products. Adv. Food Nutr. Res 2020, 91, 65–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Shanfield, H.; Zlatkis, A. Analysis of trace volatile organic compounds in coffee by headspace concentration and gas chromatography-mass spectrometry. Chromatographia 1983, 17, 411–417. [Google Scholar] [CrossRef]
- Pimenta, C.J.; Angélico, C.L.; Chalfoun, S.M. Challengs in coffee quality: Cultural, chemical and microbiological aspects. Ciênc. Agrotec. 2018, 42, 337–349. [Google Scholar] [CrossRef]
- Sharma, H. A detail chemistry of coffee and its analysis. In Coffee: Production and Research; Castanheira, D.T., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Risticevic, S.; Carasek, E.; Pawliszyn, J. Headspace solid-phase microextraction–gas chromatographic–time-of-flight mass spectrometric methodology for geographical origin verification of coffee. Anal. Chim. Acta 2008, 617, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Tripaldi, P.; Pérez-González, A.; Duchowicz, P.R.; Pis Diez, R. A retention index-based QSPR model for the quality control of rice. J. Cereal Sci. 2018, 79, 303–310. [Google Scholar] [CrossRef]
- OECD. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationships [(Q)SAR] Models; OECD: Paris, France, 2007. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvascience. alvaMolecule (Software to View and Prepare Chemical Datasets) Version 1.0.4. 2020. Available online: https://www.alvascience.com/alvamolecule/ (accessed on 1 November 2021).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millam, J.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.; Martin, R.; Morokuma, K.; Farkas, O.; Foresman, J.; Fox, D. Gaussian 16, Rev. C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yılmaz, Z.T.; Odabaşoğlu, H.Y.; Şenel, P.; Adımcılar, V.; Erdoğan, T.; Özdemir, A.; Gölcü, A.; Odabaşoğlu, M. A novel 3-((5-methylpyridin-2-yl) amino) isobenzofuran-1 (3H)-one: Molecular structure describe, X-ray diffractions and DFT calculations, antioxidant activity, DNA binding and molecular docking studies. J. Mol. Struct. 2020, 1205, 127585. [Google Scholar] [CrossRef]
- Chandran, K.; Seetharamiah, N.K.; Sambanthan, M.; Anandhan, M.; Venkatesan, R. Crystal Structure, Spectral investigations, DFT and Antimicrobial activity of Brucinium Benzilate (BBA). J. Mol. Model. 2021, in press. [Google Scholar] [CrossRef]
- Alvascience. alvaDesc (Software for Molecular Descriptors Calculation) Version 2.0.10. 2021. Available online: https://www.alvascience.com/alvadesc/ (accessed on 1 November 2021).
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; WILEY-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Ballabio, D.; Consonni, V.; Mauri, A.; Claeys-Bruno, M.; Sergent, M.; Todeschini, R. A novel variable reduction method adapted from space-filling designs. Chemom. Intell. Lab. Syst. 2014, 136, 147–154. [Google Scholar] [CrossRef]
- Leardi, R. Genetic algorithms in chemistry. In Comprehensive Chemometrics: Chemical and Biochemical Data Analysis, 2nd ed.; Brown, S., Tauler, R., Walczak, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 1, pp. 617–634. [Google Scholar]
- Alvascience. alvaModel (Software to Model QSAR Data) Version 2.0.2. 2021. Available online: https://www.alvascience.com/alvamodel/ (accessed on 1 November 2021).
- Rojas, C.; Duchowicz, P.R.; Tripaldi, P.; Pis Diez, R. QSPR analysis for the retention index of flavors and fragrances on a OV-101 column. Chemom. Intell. Lab. Syst. 2015, 140, 126–132. [Google Scholar] [CrossRef]
- Varmuza, K.; Filzmoser, P. Introduction to Multivariate Statistical Analysis in Chemometrics; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, C.; Duchowicz, P.R.; Castro, E.A. Foodinformatics: Quantitative structure-property relationship modeling of volatile organic compounds in peppers. J. Food Sci. 2019, 84, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cao, C.; Li, Z. Approach to estimation and prediction for normal boiling point (NBP) of alkanes based on a novel molecular distance-edge (MDE) vector, λ. J. Chem. Inf. Comput. Sci. 1998, 38, 387–394. [Google Scholar] [CrossRef]
- Rojas, C.; Aranda, J.F.; Jaramillo, E.P.; Losilla, I.; Tripaldi, P.; Duchowicz, P.R.; Castro, E.A. Foodinformatic prediction of the retention time of pesticide residues detected in fruits and vegetables using UHPLC/ESI Q-Orbitrap. Food Chem. 2021, 342, 128354. [Google Scholar] [CrossRef] [PubMed]
- Schuur, J.H.; Selzer, P.; Gasteiger, J. The coding of the three-dimensional structure of molecules by molecular transforms and its application to structure-spectra correlations and studies of biological activity. J. Chem. Inf. Comput. Sci. 1996, 36, 334–344. [Google Scholar] [CrossRef]
- Devinyak, O.; Havrylyuk, D.; Lesyk, R. 3D-MoRSE descriptors explained. J. Mol. Graph. Model. 2014, 54, 194–203. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, C.; Alcívar León, C.D.; Contreras Aguilar, E.; Mazón Ayala, P.V.; Muñoz, D. Quantitative Structure–Property Relationship for the Retention Index of Volatile and Semi-Volatile Compounds of Coffee. Chem. Proc. 2022, 8, 48. https://doi.org/10.3390/ecsoc-25-11731
Rojas C, Alcívar León CD, Contreras Aguilar E, Mazón Ayala PV, Muñoz D. Quantitative Structure–Property Relationship for the Retention Index of Volatile and Semi-Volatile Compounds of Coffee. Chemistry Proceedings. 2022; 8(1):48. https://doi.org/10.3390/ecsoc-25-11731
Chicago/Turabian StyleRojas, Cristian, Christian D. Alcívar León, Elizabeth Contreras Aguilar, Paola V. Mazón Ayala, and Doménica Muñoz. 2022. "Quantitative Structure–Property Relationship for the Retention Index of Volatile and Semi-Volatile Compounds of Coffee" Chemistry Proceedings 8, no. 1: 48. https://doi.org/10.3390/ecsoc-25-11731