
In Silico Evaluation of New Fluoroquinolones as Possible Inhibitors of Bacterial Gyrases in Resistant Gram-Negative Pathogens †



Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Ligands
2.2. Search for Protein Structures
2.3. Molecular Docking
2.4. Multiple Sequence Alignment
2.5. Retrospective Docking
3. Results and Discussion
3.1. General Characteristics of the Ligands
3.2. Alignment of GyrA Protein Structures
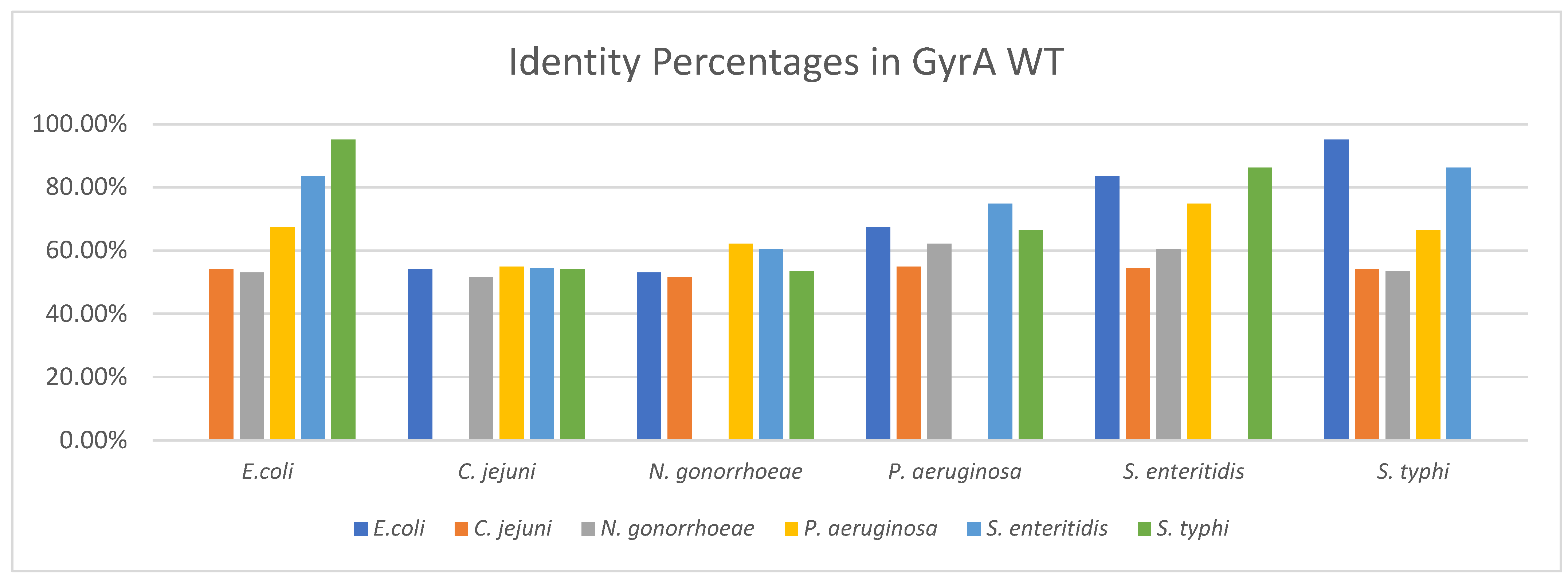
3.2.1. Identity between GyrA WT Structures
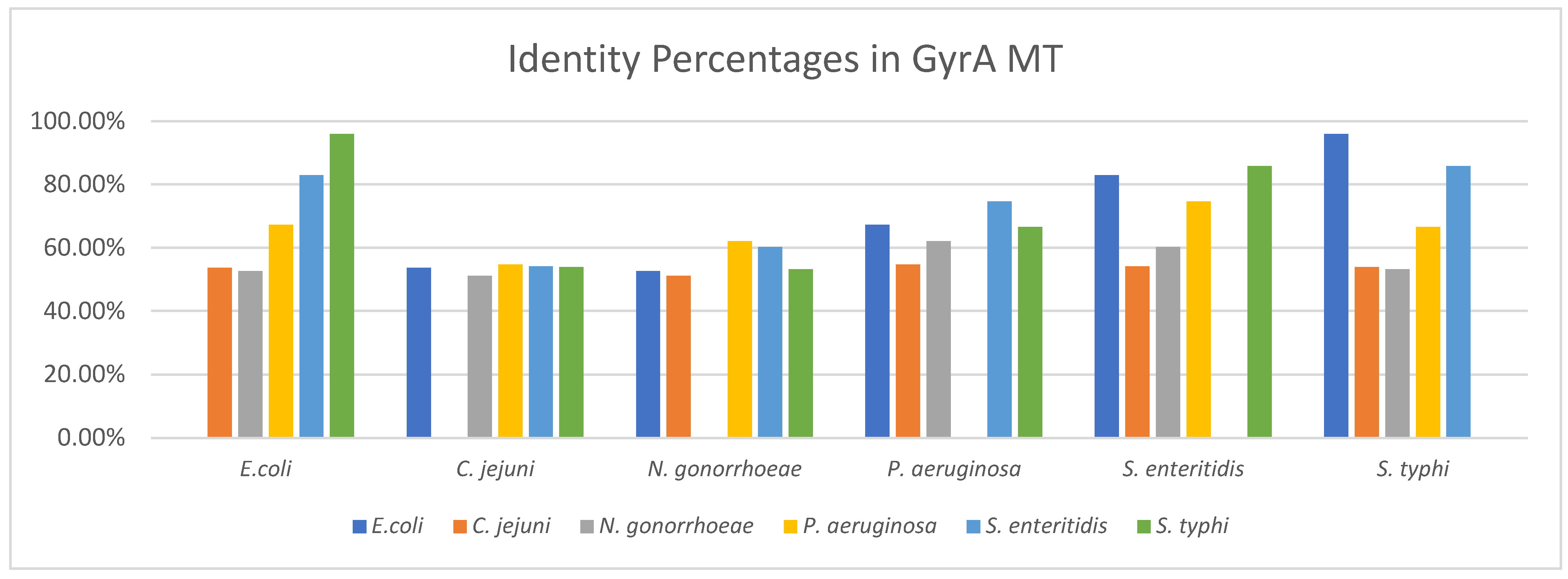
3.2.2. Identity between GyrA MT Structures
3.3. Molecular Docking
3.3.1. Molecular Docking Results in GyrA MT of Campylobacter jejuni
3.3.2. Summary of Molecular Docking Results
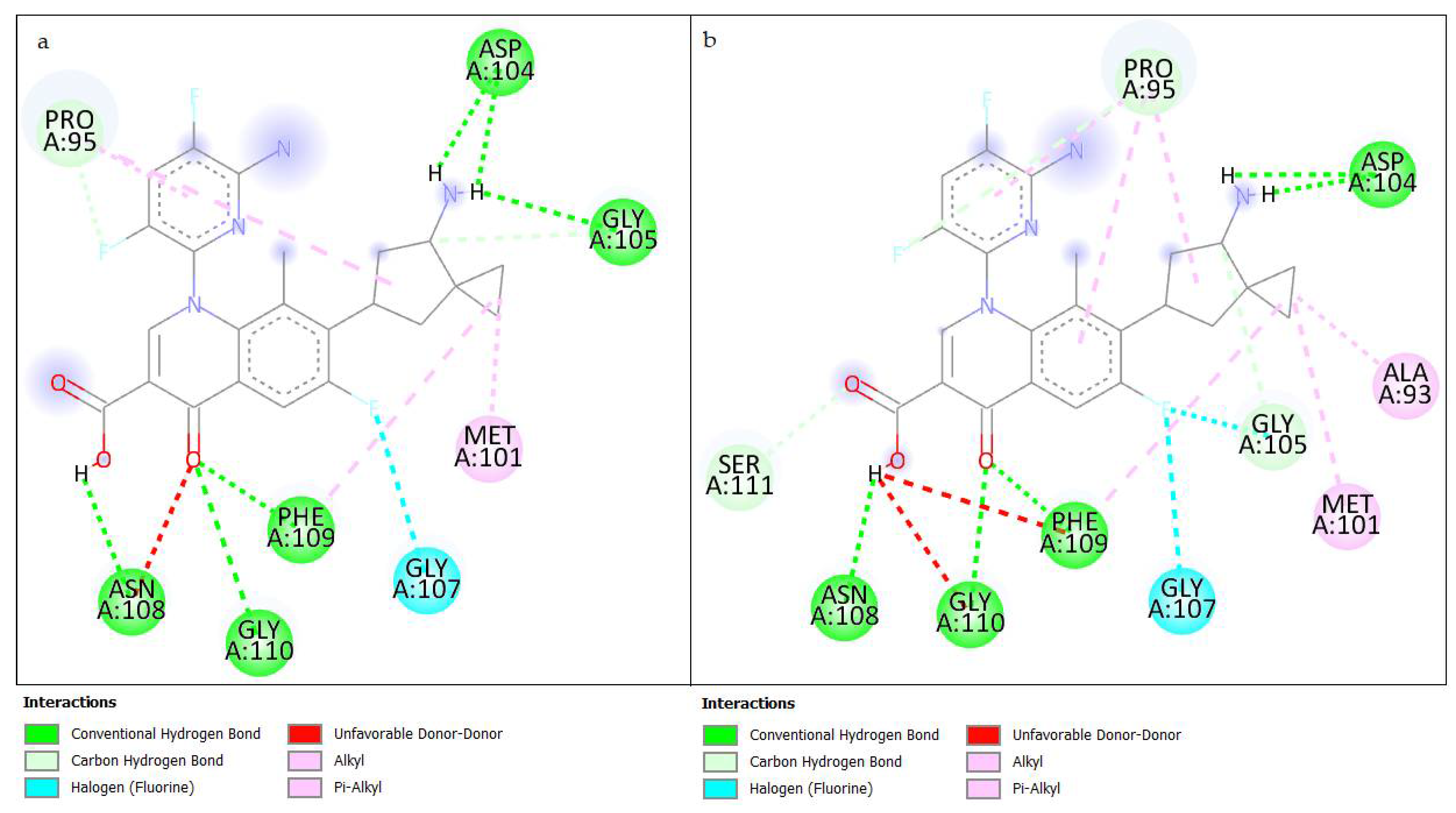
3.3.3. Results of Interactions in Molecular Docking
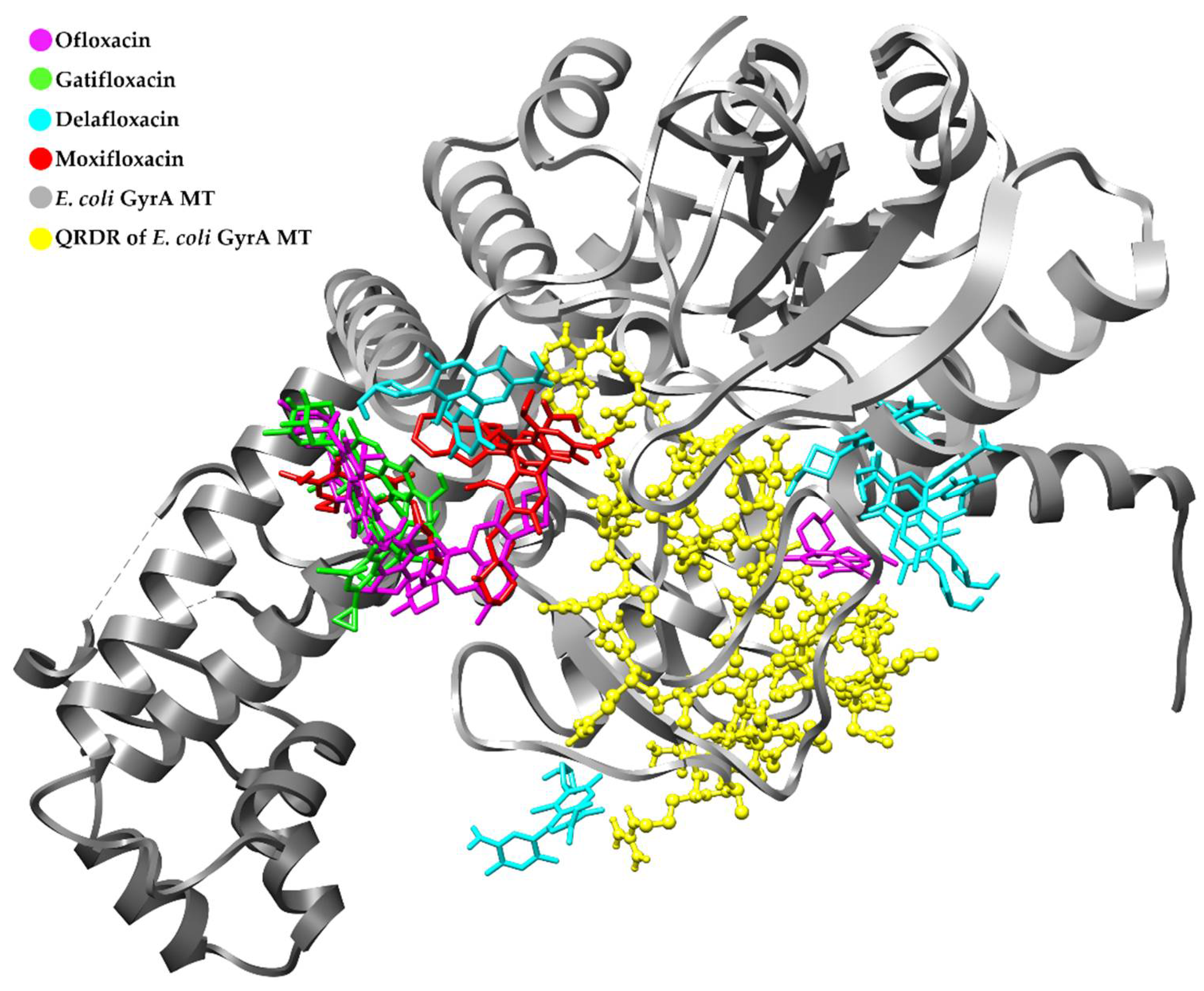
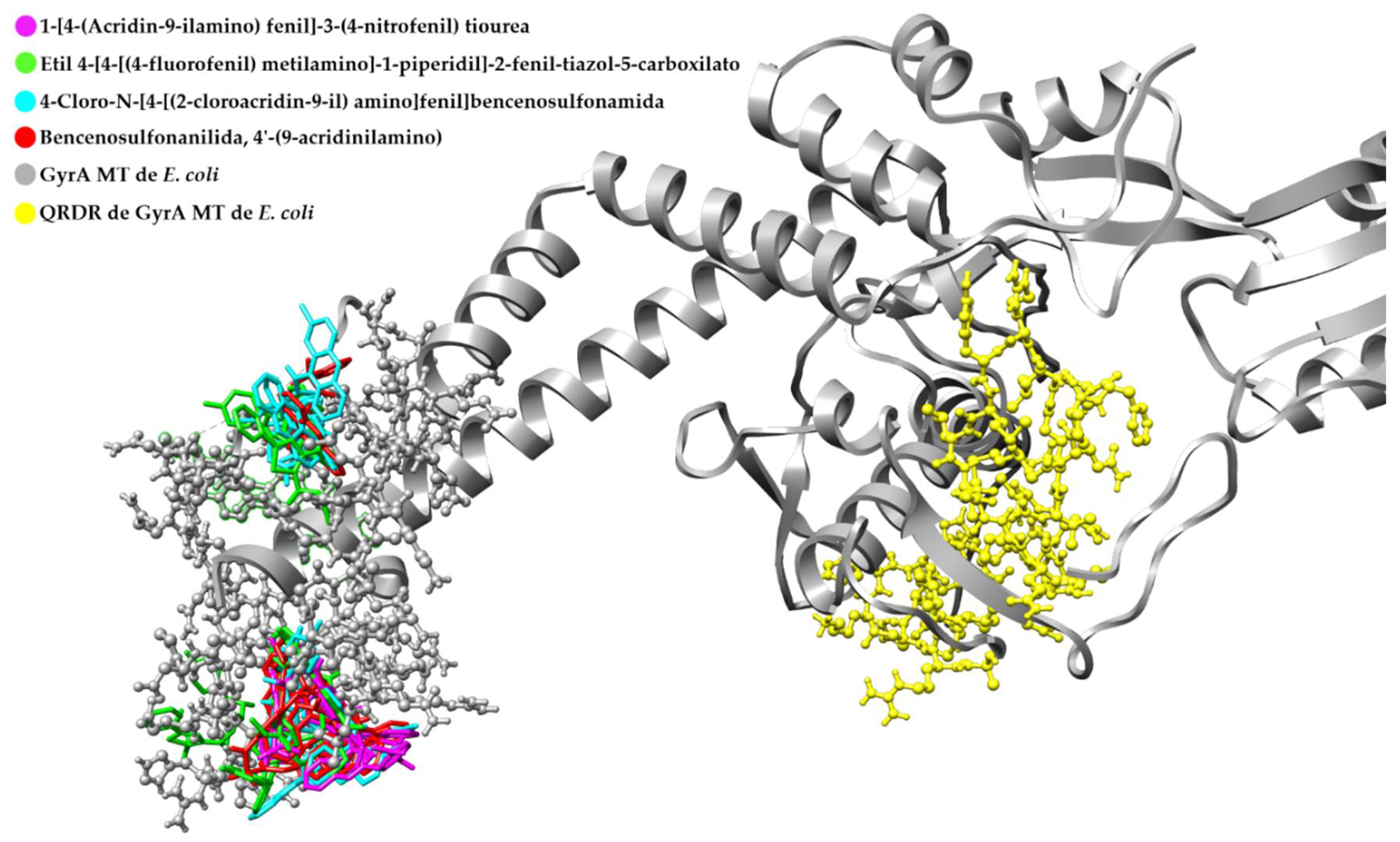
3.4. Retrospective Docking
Retrospective Docking in GyrA MT of E. coli
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brar, R.K.; Jyoti, U.; Patil, R.K.; Patil, H.C. Fluoroquinolone antibiotics: An overview. Adesh. Univ. J. Med. Sci. Res. 2020, 2, 26–30. [Google Scholar] [CrossRef]
- Mohammed, H.H.H.; Abuo-Rahma, G.E.-D.A.A.; Abbas, S.H.; Abdelhafez, E.-S.M.N. Current Trends and Future Directions of Fluoroquinolones. Curr. Med. Chem. 2019, 26, 3132–3149. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Fluit, A.C.; Schmitz, F.-J. Fluoroquinolones: Structure and Target Sites. Curr. Drug Targets 2003, 4, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Arés, F.; Martínez de la Ossa, R.; Alfayate, S. Quinolonas en Pediatría. Rev. Pediatría Atención Primaria 2017, 19, e83–e92. [Google Scholar]
- Resistencia a los Antibióticos. Available online: http://www.who.int/es/news-room/fact-sheets/detail/resistencia-a-los-antibióticos (accessed on 15 July 2021).
- Resistencia a los Antimicrobianos. Available online: https://www.paho.org/es/documentos/magnitud-tendencias-resistencia-antimicrobianos-latinoamerica-relavra-2014-2015-2016 (accessed on 15 July 2021).
- La OMS Publica la Lista de las Bacterias para las que se Necesitan Urgentemente Nuevos Antibióticos. Available online: https://www.who.int/es/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 15 July 2021).
- Ballón, W.; Grados, R. Acomplamiento molecular: Criterios prácticos para la selección de ligandos biológicamente activos e identificación de nuevos blancos terapéuticos. Rev. Con-Cienc. 2019, 7, 55–72. [Google Scholar]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Serra, H.A. Quinolonas. Separata 2008, 16, 1–9. [Google Scholar]
- Bakken, J.S. The Fluoroquinolones: How Long Will Their Utility Last? Scand J. Infect. Dis. 2004, 36, 85–92. [Google Scholar] [CrossRef]
- Heddle, J.G.; Barnard, F.M.; Wentzell, L.M.; Maxwell, A. The Interaction of Drugs with DNA Gyrase: A Model for the Molecular Basis of Quinolone Action. Nucleosides Nucleotides Nucleic Acids 2000, 19, 1249–1264. [Google Scholar] [CrossRef]
- Pierce, B.A. Génetica. un Enfoque Conceptual, 3rd ed.; Editorial Médica Panamericana: Madrid, Spain, 2009; p. 730. [Google Scholar]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharm. 2018, 103, 923–938. [Google Scholar] [CrossRef]
- Madurga, S.; Sánchez, J.; Belda, I.; Vila, J.; Giralt, E. Mechanism of Binding of Fluoroquinolones to the Quinolone Resistance-Determining Region of DNA Gyrase: Towards an Understanding of the Molecular Basis of Quinolone Resistance. ChemBioChem 2008, 9, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M. Mecanismos de resistencia a quinolonas mediada por plásmidos. Enferm. Infecc. Microbiol. Clin. 2005, 23, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Jacho, M. Métodos Computacionales para el Diseño de Fluoroquinolonas con Alta Afinidad por Topoisomerasa II de Leishmania. Bachelor’s Thesis, Universidad Central del Ecuador, Quito, Ecuador, 2020. [Google Scholar]
- Varughese, L.R.; Rajpoot, M.; Goyal, S.; Mehra, R.; Chhokar, V.; Beniwal, V. Analytical profiling of mutations in quinolone resistance determining region of gyrA gene among UPEC. PLoS ONE 2018, 13, e0190729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Wang, S.; Li, X.; He, X.; Jian, L. Development of in vitro resistance to fluoroquinolones in Pseudomonas aeruginosa. Antimicrob. Resist. Infect. Control. 2020, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nouri, R.; Ahangarzadeh, M.; Hasani, A.; Aghazadeh, M.; Asgharzadeh, M. The role of gyrA and parC mutations in fluoroquinolones-resistant Pseudomonas aeruginosa isolates from Iran. Braz. J. Microbiol 2016, 47, 925–930. [Google Scholar] [CrossRef] [Green Version]
- Hakanen, A.; Jalava, J.; Kotilainen, P.; Jousimies-Somer, H.; Siitonen, A.; Huovinen, P. gyrA Polymorphism in Campylobacter jejuni: Detection of gyrA Mutations in 162 C. jejuni Isolates by Single-Strand Conformation Polymorphism and DNA Sequencing. Antimicrob. Agents Chemother. 2002, 46, 2644–2647. [Google Scholar] [CrossRef] [Green Version]
- Paravisi, M.; Laviniki, V.; Bassani, J.; KKunert Filho, H.; Carvalho, D.; Wilsmann, D.; Borges, K.; Furian, T.; Salle, C.; Moraes, H.; et al. Antimicrobial Resistance in Campylobacter jejuni Isolated from Brazilian Poultry Slaughterhouses. Braz. J. Poult. Sci. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Da Silva, B.; Medeiros, V.; Barbosa, A.V.; Silva, W.; Faccini, F.; Lima da Costa, D.; Clementino, M.; Cosendey, M. Detection of fluoroquinolone resistance by mutation in gyrA gene of Campylobacter spp. isolates from broiler and laying (Gallus gallus domesticus) hens, from Rio de Janeiro State, Brazil. Ciencia Rural 2015, 45, 2013–2018. [Google Scholar] [CrossRef] [Green Version]
- Kivata, M.W.; Mbuchi, M.; Eyase, F.L.; Bulimo, W.D.; Kyanya, C.K.; Oundo, V.; Muriithi, S.W.; Andagalu, B.; Mbinda, W.M.; Soge, O.O.; et al. GyrA and parC mutations in fluoroquinolone-resistant Neisseria gonorrhoeae isolates from Kenya. BMC Microbiol. 2019, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Dahiya, S.; Sharma, P.; Sharma, S.; Singh, T.P.; Kapil, A.; Kaur, P. Structure Based In Silico Analysis of Quinolone Resistance in Clinical Isolates of Salmonella Typhi from India. PLoS ONE 2015, 10, e0126560. [Google Scholar] [CrossRef]
- Britto, C.D.; Dyson, Z.A.; Mathias, S.; Bosco, A.; Dougan, G.; Jose, S.; Nagaraj, S.; Holt, K.E.; Pollard, A.J. Persistent circulation of a fluoroquinolone-resistant Salmonella enterica Typhi clone in the Indian subcontinent. J. Antimicrob. Chemother. 2020, 75, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, S.; An, R.; Rendahl, A. Molecular and physiological characterization of fluoroquinolone-highly resistant salmonella enteritidis strains. Front. Microbiol 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, M.; Xu, X.; Fu, Y.; Xiong, Z.; Zhang, L.; Qu, X.; Zhang, H.; Wei, Y.; Zhan, Z.; et al. High-levels of resistance to quinolone and cephalosporin antibiotics in MDR-ACSSuT Salmonella enterica serovar Enteritidis mainly isolated from patients and foods in Shanghai, China. Int. J. Food Microbiol. 2018, 286, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.; Zhang, J.; Xu, X.; Shi, W.; Chen, S.; Yang, X.; Su, X.; Shi, X.; Meng, J. Emerging high-level ciprofloxacin resistance and molecular basis of resistance in Salmonella enterica from humans, food and animals. Int. J. Food Microbiol. 2018, 280, 1–9. [Google Scholar] [CrossRef]
- Davila, C.; Llach, L.; Salgado-Moran, G.; Ramirez-Tagle, R. Docking studies on novel analogs of quinolones against DNA gyrase of Escherichia coli. J. Pharm. Pharm. Res. 2018, 6, 386–391. [Google Scholar]
- Pacheco, E. Diseño In Silico de una Molécula con Propiedades Germicidas Inhibidora de la ADN Girasa de Pseudomonas Aeruginosa. Bachelor’s Thesis, Universidad de San Carlos de Guatemala, Guatemala City, Guatemala, 2017. [Google Scholar]
- Kampranis, S.C.; Maxwell, A. The DNA gyrase-quinolone complex. ATP hydrolysis and the mechanism of DNA cleavage. J. Biol. Chem. 1998, 273, 22615–22626. [Google Scholar] [CrossRef] [Green Version]
- Susanta, S.; Surendra, P.; Ashish, P. In-silico identification and molecular docking studies of quinolone resistance determining region (QRDR) of E. coli DNA gyrase-a with ofloxacin schiff bases. Int. J. PharmTech Res. 2013, 5, 1794–1803. [Google Scholar]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [Green Version]
- Vashist, J.; Vishvanath; Kapoor, R.; Kapil, A.; Yennamalli, R.; Subbarao, N.; Rajeswari, M.R. Interaction of nalidixic acid and ciprofloxacin with wild type and mutated quinolone-resistance-determining region of DNA gyrase A. Indian J. Biochem. Biophys. 2009, 46, 147–153. [Google Scholar]
- Medapi, B.; Meda, N.; Kulkarni, P.; Yogeeswari, P.; Sriram, D. Development of acridine derivatives as selective Mycobacterium tuberculosis DNA gyrase inhibitors. Bioorg. Med. Chem. 2016, 24, 877–885. [Google Scholar] [CrossRef]
- Cramariuc, O.; Rog, T.; Javanainen, M.; Monticelli, L.; Polishchuk, A.V.; Vattulainen, I. Mechanism for translocation of fluoroquinolones across lipid membranes. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2563–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orero, A.; Cantón, E.; Pemán, J.; Gobernado, M. Penetración de los antibióticos de los polimorfonucleares humanos, con especial referencia a las quinolonas. Rev. Española Quim. 2002, 15, 1–14. [Google Scholar]
- Vaden, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935–4947. [Google Scholar]
- Brown, S. Bioinformatics. A Biologist’s Guide to Biocomputing and the Internet; Eaton Publishing: New York, NY, USA, 2000. [Google Scholar]
- Homologia. Available online: http://webs.ucm.es/info/biomol2/bioquimicaI/WTA/Homologia.html (accessed on 7 September 2021).
- Ochman, H. Evolución y Enfermedad: ¿Qué convirtió la “salmonella” en maléfica? Mètode Sci. Stud. J. 2014, 4, 178–180. [Google Scholar]
- Contreras, B.; Yruela, I. Alineamiento de Secuencias, Estructura Secundaria, Desorden y Filogenias de Proteínas Didactic Material. Available online: http://hdl.handle.net/10261/117608 (accessed on 7 September 2021).
- Rodríguez, E. Alineamiento de Pares de Secuencias; Instituto Politécnico Nacional: Mexico City, Mexico, 2013. [Google Scholar]
- Biopolimeros. Available online: http://facultatciencies.uib.cat/prof/josefa.donoso/campus/modulos/modulo4/modulo4_13.htm (accessed on 12 July 2021).
- Aykul, S.; Martinez, E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Anal. Biochem. 2017, 508, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Beltrán, Y.; Rojas, J.A.; Morales, I.; Morris, H. Acoplamiento molecular de compuestos fenólicos de Pleurotus ostreatus con proteínas del balance redox. Rev. Cuba. Química 2019, 31, 336–356. [Google Scholar]
- Vallejo, D. Interacciones Eléctricas en Macromoléculas de Interés Biológico. Ph.D. Thesis, Universidad Nacional de La Plata, La Plata, Argentina, 2005. [Google Scholar]
- Villacis, N. Estudios Bioinformáticos Sobre la Proteína Spike del SARS-CoV-2 para el Desarrollo de Posibles Inhibidores. Bachelor’s Thesis, Universidad Técnica de Ambato, Ambato, Ecuador, 2021. [Google Scholar]
- Spinosa, M.A. Caracterización Estructural y Docking Molecular en el Receptor Cannabinoide 2. Bachelor’s Thesis, Universidad de Buenos Aires, Buenos Aires, Argentina, 2017. [Google Scholar]
- Heinzelmann, G.; Gilson, M.K. Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Baldiris, R.; Caicedo, D.; Velásquez, M.; Valdiris, V.; Vivas-Reyes, R. Docking molecular de inhibidores de actividad quinasa: Inhibición de la piridoxal quisana. Cienc Salud Virtual 2014, 6, 99–105. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular Docking in Modern Drug Discovery: In Principles and Recent Applications. In Drug Discovery and Development—New Advances; Gaitonde, V., Karmakar, P., Trivedi, A., Eds.; IntechOpen: London, UK, 2020; pp. 1–21. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | pKa | Total Energy (kcal/mol) |
|---|---|---|
| 1 | 5.42; 6.96 | −77.204 |
| 2 | 5.20; 6.94 | −94.002 |
| 3 | 5.22; 10.01 | −81.015 |
| 4 | 5.13; 9.62 | −61.277 |
| 5 | 5.22; 10.01 | −106.831 |
| 6 | 5.41; 14.76 | −99.272 |
| 7 | 5.17; 10.00 | −74.914 |
| 8 | 5.80; 14.77 | −97.200 |
| 9 | 5.45; 9.56 | −32.860 |
| Binding Free Energy (kcal/mol) | Inhibition Constant (µM) | Total Intermolecular Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | |
|---|---|---|---|---|
| Min. | −7.14 | 5.82 | −8.93 | −2.2 |
| Max. | −5.06 | 194.14 | −6.26 | −0.32 |
| R | 2.08 | 188.32 | 2.67 | 1.88 |
| R/5 | 0.416 | 37.664 | 0.534 | 0.376 |
| Threshold Value | −6.724 | 43.484 | −8.396 | −1.824 |
| Molecule | Microorganism | Binding Free Energy (kcal/mol) | Inhibition Constant (µM) | Total Intermolecular Energy (kcal/mol) | Electrostatic Energy (kcal/mol) |
|---|---|---|---|---|---|
| 7 | C. jejuni MT | −7.14 | 5.82 | −8.93 | −1.22 |
| CPX | −6.32 | 23.29 | −7.22 | −0.84 | |
| 7 | C. jejuni WT | −7.64 | 2.51 | −9.43 | −0.83 |
| CPX | −6.2 | 28.52 | −7.1 | −1.64 | |
| 7 | E. coli MT | −7.45 | 3.44 | −9.24 | −1.3 |
| CPX | −6.46 | 18.53 | −7.35 | −1.09 | |
| 7 | E. coli WT | −7.44 | 3.54 | −9.23 | −1.24 |
| CPX | −6.29 | 24.65 | −7.18 | −0.94 | |
| 3 | N. gonorrhoeae MT | −6.81 | 10.21 | −8.3 | −2.53 |
| CPX | −6.52 | 16.51 | −7.42 | −1.53 | |
| 7 | N. gonorrhoeae WT | −7.11 | 6.14 | −8.9 | −1.74 |
| CPX | −6.15 | 31.21 | −7.04 | −1.86 | |
| 7 | P. aeruginosa MT | −8.13 | 1.1 | −9.92 | −1.51 |
| CPX | −6.38 | 21.22 | −7.27 | −1.54 | |
| 7 | P. aeruginosa WT | −7.95 | 1.48 | −9.74 | −1.43 |
| CPX | −6.5 | 17.13 | −7.4 | −1.46 | |
| 5 | S. enteritidis MT | −7.91 | 1.58 | −9.11 | −0.99 |
| CPX | −6.49 | 17.56 | −7.38 | −0.93 | |
| 5 | S. enteritidis WT | −7.75 | 2.1 | −8.94 | −0.87 |
| CPX | −6.43 | 19.44 | −7.32 | −1.3 | |
| 7 | S. typhi MT | −7.38 | 3.92 | −9.17 | −1.2 |
| CPX | −6.28 | 24.96 | −7.17 | −0.93 | |
| 7 | S. typhi WT | −7.38 | 3.89 | −9.17 | −1.27 |
| CPX | −6.31 | 23.84 | −7.2 | −0.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coba-Males, M.A.; Santamaría-Aguirre, J.; Alcívar-León, C.D. In Silico Evaluation of New Fluoroquinolones as Possible Inhibitors of Bacterial Gyrases in Resistant Gram-Negative Pathogens. Chem. Proc. 2022, 8, 43. https://doi.org/10.3390/ecsoc-25-11753
Coba-Males MA, Santamaría-Aguirre J, Alcívar-León CD. In Silico Evaluation of New Fluoroquinolones as Possible Inhibitors of Bacterial Gyrases in Resistant Gram-Negative Pathogens. Chemistry Proceedings. 2022; 8(1):43. https://doi.org/10.3390/ecsoc-25-11753
Chicago/Turabian StyleCoba-Males, Manuel Alejandro, Javier Santamaría-Aguirre, and Christian D. Alcívar-León. 2022. "In Silico Evaluation of New Fluoroquinolones as Possible Inhibitors of Bacterial Gyrases in Resistant Gram-Negative Pathogens" Chemistry Proceedings 8, no. 1: 43. https://doi.org/10.3390/ecsoc-25-11753