
A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles †


Department of Chemistry, Maharaja Bir Bikram College, Agartala 799004, India
†
Presented at the 25th International Electronic Conference on Synthetic Organic Chemistry, 15–30 November 2021; Available online: https://ecsoc-25.sciforum.net/ .
Chem. Proc. 2022, 8(1), 27; https://doi.org/10.3390/ecsoc-25-11726
Published: 14 November 2021
(This article belongs to the Proceedings of The 25th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:N-Arylamides are a ubiquitous component of a broad range of natural products and biologically active compounds. In this paper, a new synthetic protocol for the preparation of N-arylamides was developed via the hypervalent iodine-mediated aza-Hofmann-type rearrangement of amidines. The reaction proceeded smoothly at 100 °C in the presence of PhINTs in toluene solvent. The requisite amidine substrates were prepared from amines and nitriles by applying the Pinner reaction approach. Considering the easy access of amidines from nitriles, the overall process is the conversion of nitriles to acetanilide and N-arylamides. As an application of the protocol, the preparation of paracetamol from 4-cyanophenol is also described.
1. Introduction
Amide-bond-containing compounds are a ubiquitous component of a broad range of natural products and biologically active compounds [1,2]. In recent years, molecules with amide moieties have attracted considerable attention in medicinal chemistry due to their significant and diverse biological activities, including antipyretic [3], antimalarial [4], anti-inflammatory [5], and antitumor [6] effects. A recent study showed that about 25% of the known pharmaceuticals contain at least one amide bond [7]. More importantly, the amide bond constitutes the backbone of the crucial biological proteins and peptides. N-Arylamides are an important class of amides widely present in natural products (e.g., penicillin and paclitaxel), pharmaceuticals, and agrochemicals, as well as in a large number of industrial materials including polymers, detergents, and lubricants [8,9]. The most popular methods for the preparation of this class of compounds rely on the reaction of activated carboxylic acid derivatives, such as chlorides, anhydrides or esters, with amines or, alternatively, the direct union of the carboxylic acids with amines assisted by stoichiometric amounts of coupling reagents [10,11]. However, these classical approaches possess low atom efficiency and generate large amounts of waste products, making their environmental profile unfavorable. New synthetic approaches that do not require activation of the carboxylic acid with a stoichiometric reagent, based on a Lewis acid (e.g., boronic acids) [12] or silica [13] as a catalyst, were developed. Catalyst poisoning and substrate scope are the main challenges remaining in this attractive approach. Among the transition-metal-catalyzed synthetic methods developed so far [14], the direct formation of the C–N bond through the cross-coupling reaction of arylhalides (I, Br, Cl) or pseudohalides (OTf, OTs, OMs, etc.) with primary or secondary amides is one of the best methods in terms of versatility [15]. These amidation methods are mainly catalyzed by the transition metal such as palladium and copper catalysts; and it is necessary to install the leaving group beforehand on the aromatic coupling partner, which finally ends up as undesirable waste. It is, therefore, highly desirable to develop an efficient and more environmental friendly method for the synthesis of N-arylamides.
In recent years, hypervalent iodine compounds have emerged as environmentally friendly and efficient oxidizing reagents for various synthetically useful oxidative transformations [16]. These compounds are stable, less toxic, commercially available, and easy to handle. Currently, various hypervalent iodine reagents are widely used as green oxidants for the Hofmann rearrangement of primary amides [17]. In this context, very recently, Li and co-workers reported the Hoffmann-type rearrangement of primary amides to secondary amides using PhI(OAc)2 as an oxidizing reagent [18]. In the 1990s Ramsden and co-workers described the phenyliodine(III)diacetate (PIDA)-mediated oxidative rearrangement of N-substituted amidines to carbodiimides [19]. Recently, we observed that carbodiimides obtained from amidines can easily be transformed into acetanilides via reaction with acetic acid during the in situ generation from PhI(OAc)2 [20]. Although this method is highly efficient for the preparation of acetanilides, there are some disadvantages associated with this protocol. The main limitation of the protocol is the limited substrate scope that is restricted to the synthesis of acetanilides only. Different hypervalent iodine reagents were required for the preparation of anilides other than acetanilide, which reduces its applicability in the development of chemical projects. Therefore, we envisaged that by exploring suitable oxidant systems, the aza-Hofmann rearrangement of amidines would lead to the in situ formation of carbodiimides. The subsequent reaction of carbodiimides with a carboxylic acid may provide easy access to N-arylamides (anilides) in a one-pot process.
In this paper, we demonstrate a new synthetic protocol for the preparation of N-arylamides including paracetamol via the hypervalent iodine-mediated aza-Hofmann-type rearrangement of amidines. The requisite amidine substrates were prepared from amines and nitriles by applying the Pinner reaction approach [21]. Considering the easy access of amidines from nitriles, the overall process is the conversion of nitriles to anilides. As an application of the protocol, we synthesized paracetamol from 4-cyanophenol.
2. Materials and Methods
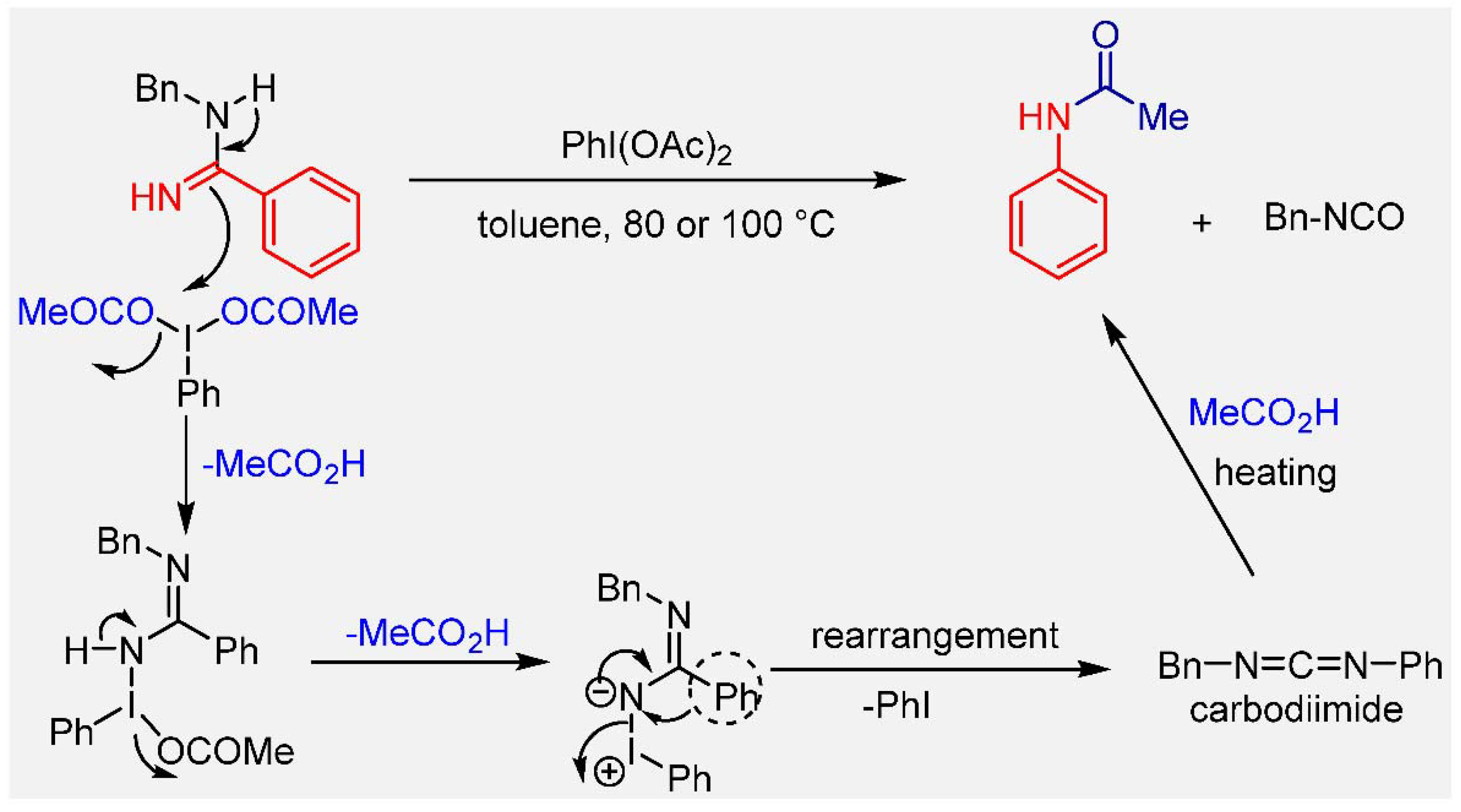
In our previous work, we demonstrated the synthesis of secondary amides from N-substituted amidines by tandem oxidative rearrangement and isocyanate elimination. In that approach, the PhI(OAc)2-mediated oxidative rearrangement of N-substituted amidines in situ generated a carbodiimide intermediate, which was subsequently trapped by an acetic acid generated in situ from PhI(OAc)2 to provide the corresponding acetanilides (Scheme 1). The main disadvantage of this protocol is that different hypervalent iodine reagents are required for the synthesis of anilides other than acetanilide, which reduces its applicability in the development of chemical projects. Therefore, we envisaged that by exploring suitable oxidant systems other than PhI(OAc)2, the oxidative rearrangement of amidines would lead to the in situ formation of carbodiimides. The subsequent reaction of carbodiimide with a carboxylic acid would provide access to anilides in one-pot process. The efficient oxidative rearrangement of amidines without the competitive nucleophilic addition of nucleophiles generated in situ may be an efficient approach for the success of this transformation. Moreover, the reactivity of hypervalent iodine can be modulated by changing the substituents, and the nucleophile generated in situ can be selected.
Based on these elegant ideas, we started our investigation with N-benzylbenzamidine (1a) as a model substrate for the generation of carbodiimide. Benzoic acid (2a) was selected for the reaction with carbodiimide. When 1a and 2a were heated with 1.5 equiv. PhI(OAc)2 in toluene at 100 °C for 15 h, a mixture of two anilides, benzanilide (3a) and acetanilide (4a), was obtained in 48% and 40% yields, respectively (Table 1, entry 1). Unfortunately, we did not obtain any product when the reaction was performed with PhI(OCOCF3) as an oxidant (Table 1, entry 2). Interestingly, by switching the hypervalent iodine reagent from PhI(OAc)2 to PhINTs, a full conversion was achieved in an overnight reaction (15 h) and only one product, benzanilide, was obtained in 86% yield (Table 1, entry 3). In this reaction, N-tosyl aniline was eliminated as a by-product from PhINTs. Due to the lower nucleophilicity of N-tosyl aniline, benzoic acid attacks the carbodiimide, leading to the formation of benzanilide product. The screening of solvent revealed that toluene is the best solvent for this transformation (entries 4–6). Next, we investigated the influence of the base on this tandem reaction. It was observed that an additive (base) either has no influence or a negative influence on the yield of product (Table 1, entries 7–10). Thus, the optimal reaction conditions for this transformation are as follows: amidine (0.5 mmol), carboxylic acid (1 mmol), PhINTs (0.75 mmol), and toluene (1 mL), at 100 °C for 15 h.
3. Results and Discussion
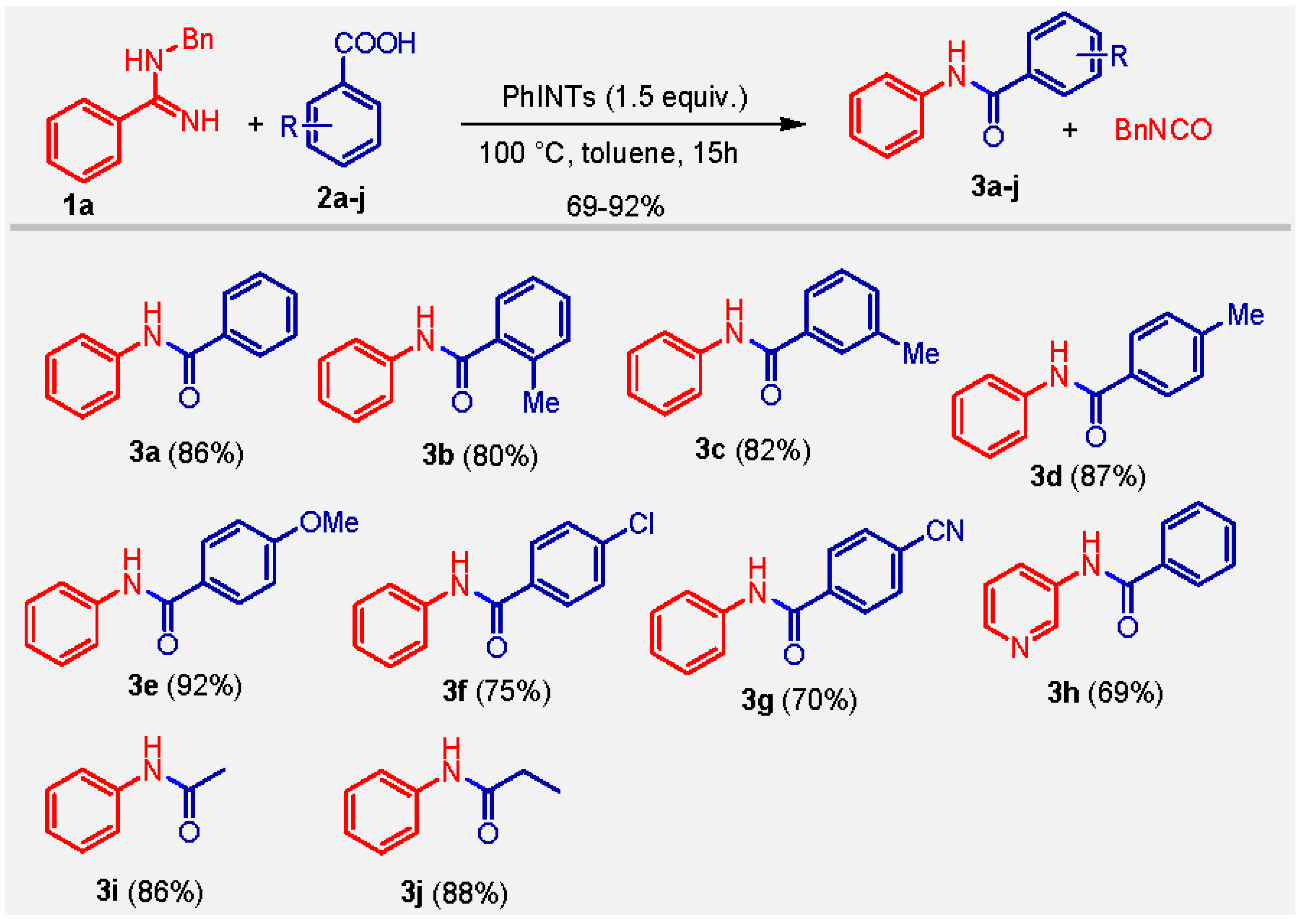
We then explored the substrate scope and limitation of the protocol. Various aliphatic and aromatic carboxylic acids bearing electron-donating as well as electron-withdrawing groups were reacted with the carbodiimide generated from N-benzylbenzamidine (1a), affording benzanilides (3) in good to excellent yields. Various functional groups, such as Me, OMe, Cl, and CN, substituted at ortho-, meta-, and para-positions of aromatic carboxylic acids were well tolerated under the reaction conditions and resulted in the desired products in high yields (Scheme 2).
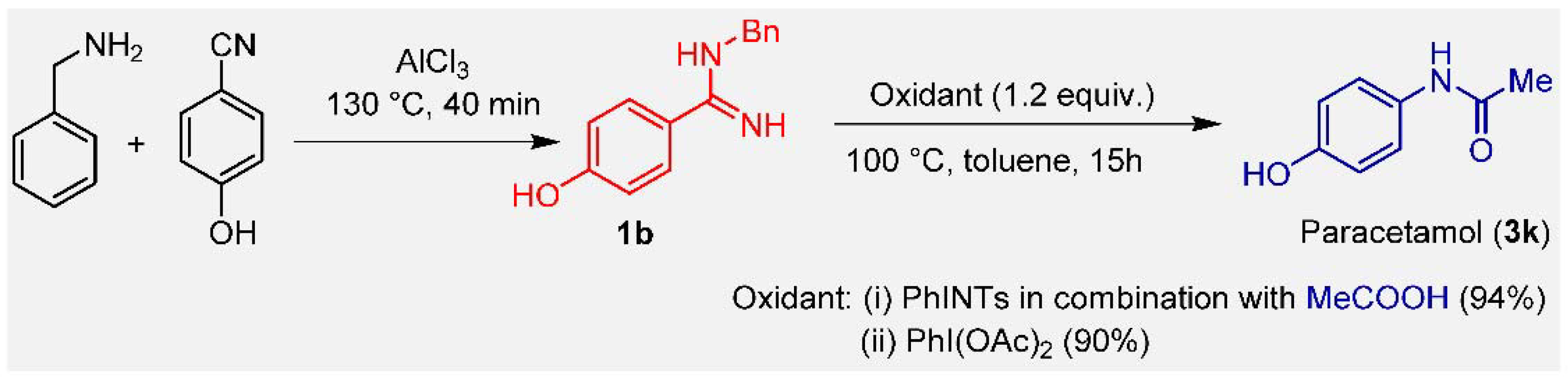
N-Acetyl-para-aminophenol (APAP), commonly known as paracetamol or acetaminophen, is a representative of the N-arylamide class drug. This drug is one of the most consumed worldwide, with a global production of more than 100,000 tons per year. Over the last century, many routes have been explored for the preparation of paracetamol [22,23,24], but all those that have emerged industrially are based on the acetylation of para-aminophenol (PAP) as the final stage [25,26]. We applied this protocol for the preparation of paracetamol starting from 4-cyanophenol. The reaction of 4-cyanophenol with benzylamine in the presence of 1.2 equiv. of anhydrous AlCl3 at 130 °C was conducted to give the corresponding amidine (1b). The oxidative rearrangement of 1b with PhI(OAc)2 (1.5 equiv.) in toluene at 100 °C for 15 h was conducted to give the paracetamol (3k) in 90% yield (Scheme 3). When the same reaction was carried out with PhINTs, 94% paracetamol was obtained. The overall process can be considered as the conversion of 4-cyanophenol into paracetamol.
4. Conclusions
In conclusion, we developed an efficient and sustainable protocol for the preparation of N-arylamides (anilides) from N-substituted amidines. The reaction proceeded smoothly with PhINTs at 100 °C in toluene solvent. Various substituted N-arylamides were obtained in high yields under oxidative reaction conditions. As an application of this protocol, we synthesized paracetamol with a high yield starting from 4-cyanophenol.
5. Experimental
General procedure for the synthesis of N-substituted amidines from amines and carbonitriles: A pressure flask (50 mL) equipped with a small stirring bar was charged with the amine (5.5 mmol, 1.1 equiv.) and the carbonitrile (5.0 mmol, 1.0 equiv). AlCl3 (0.7 g, 0.5 equiv.) was added in one portion. The flask was tightly sealed with a Teflon screw cap and placed into a preheated oil bath at 130 °C. The reaction mixture was stirred for 40 min, and subsequently taken out of the oil bath. Ice water (50 mL) was added and under vigorous stirring concentrated aqueous NaOH (2 M) was added until a pH of 14 was reached. The aqueous layer was extracted with dichloromethane (50 mL). The combined organic layers were washed with water and then brine, and dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified using chromatography with heptane/ethyl acetate-7N NH3 in MeOH (19:1) eluent.
General procedure for the PhINTs-mediated oxidative rearrangement of N-substituted amidines to anilides: In an oven-dried microwave vial (10 mL) equipped with a magnetic stirring bar, the N-benzylbenzamidine (0.5 mmol), carboxylic acid (1 mmol) and PhINTs (0.75 mmol) were charged. The vessel was flushed with N2 and then sealed with septum. A total of 1 mL of dry toluene was added to the vessel and the reaction mixture was heated at 100 °C for 15 h. After completion of the reaction, the toluene was evaporated under reduced pressure. The crude product was purified by chromatography using hexane and ethylacetate (19:1) as eluent.
N-Phenylbenzamide (3a): 1H NMR (400 MHz, DMSO-d6): δ = 7.07 (t, J = 7.4 Hz, 1H), 7.32 (t, J = 8.1 Hz, 2H), 7.48–7.56 (m, 3H), 7.75 (d, J = 7.7 Hz, 2H), 7.92 (d, J = 7.0 Hz, 2H), 10.18 (brs, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ = 120.8, 124.1, 128.1, 128.8, 129.0, 132.0, 135.5, 139.6, 166.0.
N-(4-hydroxyphenyl)acetamide (3k):1H NMR (400 MHz, DMSO-d6) δ = 1.98 (s, 3H), 6.68 (dd, J = 9.0, 2.5 Hz, 2H), 7.34 (d, J = 7.0 Hz, 2H), 9.12 (brs, 1H, OH), 9.65 (brs, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ = 24.2, 115.4, 121.2, 131.5, 153.5, 166.9 ppm.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
The author is thankful to Tripura University, Suryamaninar, Tripura, India for providing the Bruker-400 spectrometer facility for spectral analysis.
Conflicts of Interest
The author declares no conflict of interest.
References
- Humphrey, J.M.; Chamberlin, A.R. Chemical synthesis of natural product peptides: Coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 1997, 97, 2243–2266. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Sullivan, J.E.; Farrar, H.C. Fever and antipyretic use in children. Pediatrics 2011, 127, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Freund, Y.R.; Berry, P.; Ciaravino, V.; Erve, J.C.L.; Rosenthal, P.J.; et al. Benzoxaborole antimalarial agents. Part 5. Lead optimization of novel amide pyrazinyloxy benzoxaboroles and identification of a preclinical candidate. Med. Chem. 2017, 60, 5889–5908. [Google Scholar] [CrossRef] [PubMed]
- Walesa, K.G.; Stec, A.P. The synthesis and properties of N-substituted amides of 1-(5-methylthio-1,2,4-triazol-3-yl)-cyclohexane-2-carboxylic acid. Med. Acad. Lub. 2003, 9, 118–125. [Google Scholar]
- Warnecke, A.; Fichtner, I.; Sab, G.; Kratz, F. Synthesis, cleavage profile, and antitumor efficacy of an albumin-binding prodrug of methotrexate that is cleaved by plasmin and cathepsin B. Arch. Pharm. Chem. Life Sci. 2007, 340, 389–395. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Chen, Y.; Pienkowski, E.W.; Ju, J.; Lin, S.; Rajski, S.R.; Shen, B.J. Characterization of FdmV as an amide synthetase for fredericamycin A biosynthesis in Streptomyces griseus ATCC 43944. Biol. Chem. 2010, 285, 38853–38860. [Google Scholar] [CrossRef] [Green Version]
- Lakouraj, M.; Mokhtary, M.J. Synthesis of polyamides from p-Xylylene glycol and dinitriles. Polym. Res. 2009, 16, 681–686. [Google Scholar] [CrossRef]
- Bailey, P.D.; Mills, T.J.; Pettecrew, R.; Price, R.A. Comprehensive Organic Functional Group Transformations II: Carbon with No Attached Heteroatoms; Katrizky, A.R., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2005; Volume 5, p. 201. [Google Scholar]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Charville, H.; Jackson, D.; Hodges, G.; Whiting, A. The thermal and boron-catalysed direct amide formation reactions: Mechanistically understudied yet important processes. Chem. Commun. 2010, 46, 1813–1823. [Google Scholar] [CrossRef]
- Comerford, J.W.; Farmer, T.J.; MacQuarrie, D.J.; Breeden, S.W.; Clark, J.H. Mesoporous Structured Silica—An improved catalyst for direct amide synthesis and its application to continuous flow processing. Arkivoc 2012, 2012, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.L.; Williams, J.M.J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405–3415. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Buchwald, S.L. Pd-catalyzed intermolecular amidation of aryl halides: The discovery that xantphos can be trans-chelating in a palladium complex. Am. Chem. Soc. 2002, 124, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P. Recent advances in the synthesis of amides via oxime rearrangements and its applications. Curr. Org. Synth. 2018, 15, 666–706. [Google Scholar] [CrossRef]
- Wang, X.; Yang, P.; Hu, B.; Zhang, Q.; Li, D.J. Hypervalent iodine reagents promote a facile and efficient transformation of primary amides to secondary amides. Org. Chem. 2021, 86, 2820–2826. [Google Scholar] [CrossRef]
- Ramsden, C.A.; Rose, H.L. Oxidative rearrangement and cyclisation of N-substituted amidines using iodine (III) reagents and the influence of leaving group on mode of reaction. Chem. Soc. Perkin Trans. 1 1997, 2319–2327. [Google Scholar] [CrossRef]
- Debnath, P.; Baeten, M.; LefÀvre, N.; Daele, S.V.; Maes, B.U.W. Synthesis of Secondary Amides from N-Substituted Amidines by Tandem Oxidative Rearrangement and Isocyanate Elimination. Adv. Synth. Catal. 2015, 357, 197–209. [Google Scholar] [CrossRef]
- Noe, M.; Perosa, A.; Selva, M. A flexible Pinner preparation of orthoesters: The model case of trimethylorthobenzoate. Green Chem. 2013, 15, 2252–2260. [Google Scholar] [CrossRef] [Green Version]
- Caskey, D.C.; Chapman, D.W. Process for Preparing p-Aminophenol and Alkyl Substituted p-Aminophenol. U.S. Patent 4,571,437, 18 February 1986. [Google Scholar]
- Rode, C.V.; Vaidya, M.J.; Chaudhari, R.V. Synthesis of p-aminophenol by catalytic hydrogenation of nitrobenzene. Organic process research & development. Org. Process Res. Dev. 1999, 3, 465–470. [Google Scholar]
- Joncour, R.; Duguet, N.; Metay, E.; Ferreira, A.; Lemaire, M. Amidation of phenol derivatives: A direct synthesis of paracetamol (acetaminophen) from hydroquinone. Green Chem. 2014, 16, 2997–3002. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Y.; Wang, Y.; Xue, W.; Zhao, X.J. Synthesis of p-aminophenol from the hydrogenation of nitrobenzene over metal–solid acid bifunctional catalyst. Chem. Technol. Biotechnol. 2008, 83, 1466–1471. [Google Scholar] [CrossRef]
- Liu, P.; Hu, Y.; Ni, M.; You, K.; Luo, H. Liquid phase hydrogenation of nitrobenzene to para-aminophenol over Pt/ZrO 2 catalyst and SO42−/ZrO2–Al2O3 solid acid. Catal. Lett. 2010, 140, 65–68. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of secondary amides via PhI(OAc)2-mediated oxidative rearrangement of N-substituted amidines.
Scheme 1.
Synthesis of secondary amides via PhI(OAc)2-mediated oxidative rearrangement of N-substituted amidines.

Scheme 2.
PhINTs-mediated synthesis of benzanilides from N-benzylbenzamidine.

Scheme 3.
Synthesis of paracetamol from amidine.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization reactions for the preparation of benzanilide from N-benzylbenzamidine.

| Entry | Oxidant (1.5 eq) | Solvent (1 mL) | Additives (1.1 eq) | Yield (%) 3a/4a |
|---|---|---|---|---|
| 1 | PhI(OAc)2 | Toluene | - | 48/40 |
| 2 | PhI(OCOCF3) | Toluene | - | 0/0 |
| 3 | PhINTs | Toluene | - | 86/0 |
| 4 | PhINTs | THF | - | 74/0 |
| 5 | PhINTs | DMF | - | 35/0 |
| 6 | PhINTs | o-Xylene | - | 68/0 |
| 7 | PhINTs | Toluene | Et3N | 77/0 |
| 8 | PhINTs | Toluene | AcOK | 58/34 |
| 9 | PhINTs | Toluene | Cs2CO3 | 42/0 |
| 10 | PhINTs | Toluene | Pyridine | 58/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Debnath, P. A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles. Chem. Proc. 2022, 8, 27. https://doi.org/10.3390/ecsoc-25-11726
AMA Style
Debnath P. A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles. Chemistry Proceedings. 2022; 8(1):27. https://doi.org/10.3390/ecsoc-25-11726
Chicago/Turabian StyleDebnath, Pradip. 2022. "A New Approach for the Synthesis of N-Arylamides Starting from Benzonitriles" Chemistry Proceedings 8, no. 1: 27. https://doi.org/10.3390/ecsoc-25-11726