1. Plenary Presentations
1.1. Addressing Underexplored Anti-Infective Targets
Anna K. H. Hirsch 1
1
Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research, 38124 Braunschweig, Germany
2
Department of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
The challenges associated with anti-infective drug-discovery programmes can be tackled by combining phenotypic antibacterial screening with several established and unprecedented hit-identification strategies. This approach will be illustrated using two targets: The first is a vitamin transporter from the energy-coupling factor (ECF) class, which consists of an energising module and a substrate-binding protein (S-component). Different S-components can interact with the same energising module. Here, we report on the structure-based virtual screening (SBVS), design, synthesis and structure–activity relationships (SARs) of the first class of selective, antibacterial agents against the energy-coupling factor (ECF) transporters. Having identified a druggable pocket in the crystal structure of the L. delbrueckii ECF transporter, which should play a key role in the unique mechanism of transport, our SBVS of the zinc library afforded a fragment-like hit with a good in vitro and cell-based activity and a good in vitro absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. Having established a new cell-based uptake assay in Lactobacillus casei, we identified a low-micromolar inhibitor of the ECF transporters with a broad spectrum of activity (minimum inhibitory concentration (MIC) values in the single-digit micromolar range) and a lack of resistance development.
The second is an extracellular metalloprotease and virulence factor of Pseudomonas aeruginosa, the elastase LasB. In a structure-based manner, we succeeded in optimising highly selective and potent inhibitors as promising anti-virulence agents. Multiparameter optimisation is currently ongoing based on extensive in vitro and ex vivo profiling, including the establishment of complex biological assays.
1.2. Combination Light-Based Therapies to Treat Pancreatic and Bile Duct Tumours
Pilar Acedo
Division of Medicine, Institute for Liver & Digestive Health, University College London, London NW3 2QG, UK
Over the last decades, advances in detection and treatment have revolutionised cancer medicine, and survival has improved. However, progress has not advanced equally for all types of cancers. Reduced intracellular drug accumulation coupled with multi-drug resistance is among the most common mechanisms of resistance to therapy of solid tumours. Determining the best combination of therapies for patients is a major goal, but currently major limitations in efficacy and toxicity remain. Hence, there is an urgent need to improve multimodal strategies for therapy. In this context, minimally invasive therapies capable of local tumour destruction, such as photodynamic therapy (PDT), in combination with systemic therapy, may play a potentially important role in overcoming resistance mechanisms and escape pathways. I will present different strategies that I have explored over the years to improve the therapeutic response of hard-to-treat cancers.
1.3. The Expanding Role of Prodrugs in Contemporary Drug Design and Development
Jarkko Rautio
School of Pharmacy, University of Eastern Finland, FI-70211 Kuopio, Finland
The current interest in prodrugs is evident. In the past 10 years, the Food Drug administration (FDA) has approved over 30 prodrugs, and approximately 10% of all marketed drugs worldwide can be considered to be prodrugs. Prodrug strategies are versatile and powerful tools to improve the problematic characteristics of molecules. These strategies have traditionally been embarked on to address absorption, distribution, metabolism, excretion (ADME) properties and the risks of marketed drugs or as a tool in late-stage problem-solving for drug candidates in development phases. However, prodrug design is now being integrated into early drug discovery.
From a technical and commercial point of view, prodrugs have successfully enabled the parenteral administration of several poorly soluble drugs. Similarly, prodrug strategies have also improved the solubility and dissolution rate of poorly soluble oral drugs. Most prodrugs on the market are lipophilic derivatives of their polar and/ionized parent drugs. Targeted drug action can be, on the other hand, achieved by either the site-directed delivery or site-specific activation of a prodrug.
Admittedly, embarking on a prodrug strategy can certainly present its own challenges. However, depending on the chemical nature of the parent drug and the therapeutic target, the prodrug design can often represent a comparably smaller challenge than the alternative of searching for a new therapeutically active molecule that also inherently possesses the desired absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties.
2. Oral Presentations
2.1. Novel Quaternary Ammonium Fluoroquinolone-Based Antibacterials: Synthesis, Antimicrobial Evaluation and Docking Studies
Joanna Fedorowicz 1,2,*,
Małgorzata Morawska 1,2,3,
Shella Gilbert-Girard 3,
Liliana Mazur 4,
Cristina Durante-Cruz 3,
Heidi Mäkkylä 3,
Kirsi Savijoki 3,
Adyary Fallarero 3,
Päivi Tammela 3
and
Jarosław Sączewski 2
1
Department of Chemical Technology of Drugs, Medical University of Gdańsk, 80-416 Gdańsk, Poland
2
Department of Organic Chemistry, Medical University of Gdańsk, 80-416 Gdańsk, Poland
3
Faculty of Pharmacy, University of Helsinki, 00100 Helsinki, Finland
4
Faculty of Chemistry, Maria Curie-Sklodowska University, 20-031 Lublin, Poland
Antimicrobial resistance was identified by the WHO as one of the three greatest threats to human health, a particularly serious problem for patients whose immune system is compromised. Persistent pathogens lead to higher healthcare costs due to more expensive drugs and extended hospital stays. Since a single drug is not always able to adequately control the illness, a combination of medicines with different pharmacotherapeutic profiles may be required. Hybrid drugs are molecules intended to act at multiple targets. Interestingly, the fluoroquinolone drugs linked to other antibacterial agents represent the most comprehensively described hybrid compounds [
1].
Recently, in our laboratory, the fluorescent fluoroquinolone hybrid compounds featuring a fused quaternary quinolone-triazolinium moiety have been developed [
2,
3,
4]. The novel derivatives exhibited an antibacterial activity against various pathogens, including biofilm-forming
Pseudomonas aeruginosa, featured a delayed antibiotic resistance development, caused a defect in DNA decatenation, and were potent DNA gyrase inhibitors comparable to the reference drug, ciprofloxacin.
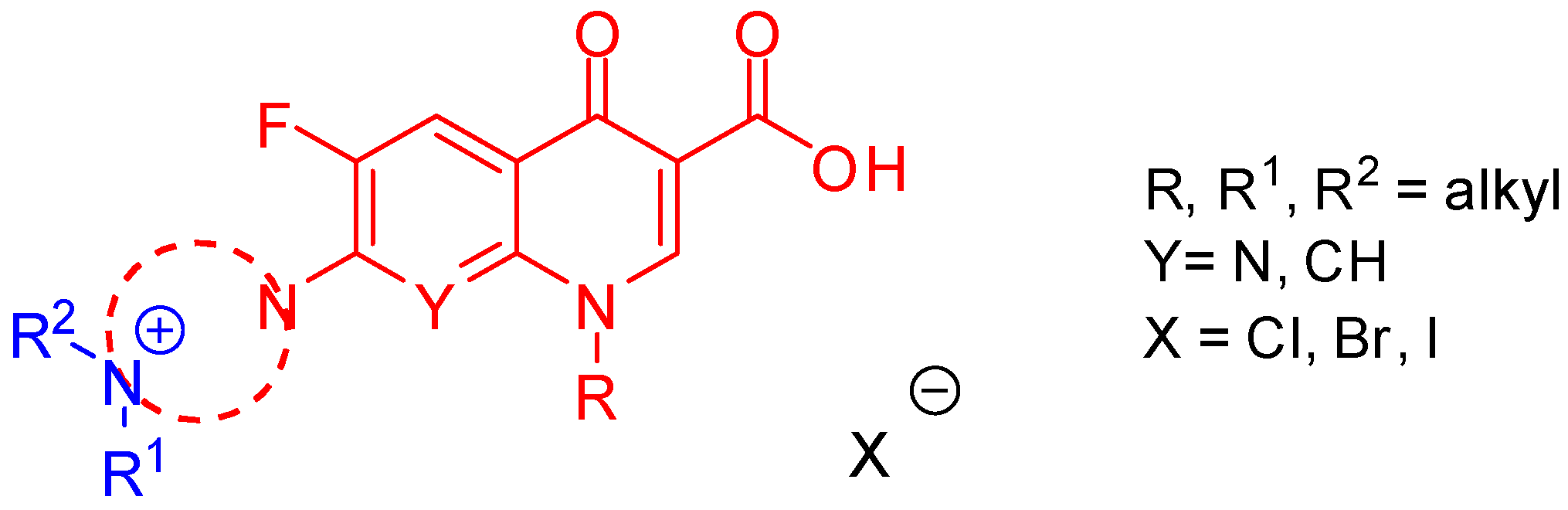
This project aimed at evaluating the biological activity of new antibacterials incorporating a fluoroquinolone drug and a quaternary ammonium compound (
Figure 1) to confirm the hypothesis that a new class of hybrid agents exhibits an unique dual mechanism of action: the destabilization of bacterial membrane structures due to the presence of a quaternary nitrogen atom and the inhibition of bacterial type II topoisomerases elicited by the fluoroquinolone portion.
The novel fluoroquinolone derivatives were designed and synthesized by an exhaustive alkylation reaction to obtain compounds incorporating a permanent positive charge on the nitrogen atom. The products were characterized by NMR, IR, MS, X-ray crystallography, and elemental analysis. Subsequently, the obtained derivatives were screened in vitro for antimicrobial activity against a panel of Gram-positive and Gram-negative bacterial strains. The most active compounds exhibited a pronounced antibacterial action in the low micro- and nanomolar range, especially towards pathogens from the ESKAPE group. Molecular docking experiments revealed that all the synthesized compounds were able to interact with active sites of bacterial type II topoisomerases pursuant to the fluoroquinolone-binding mode.
Acknowledgments
The project was financed by the Polish National Agency for Academic Exchange as part of the Bekker Scholarship Programme and subsidies of the Medical University of Gdańsk.
2.2. Small Organic Molecules to Trigger the Activity of NK Cells
Pedro F. Pinheiro 1,*,
Gonçalo C. Justino 1,
Joana P. Miranda 2
and
M. Matilde Marques 1
1
Centro de Química Estrutural—Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal
2
Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, 1649-003 Lisbon, Portugal
3
Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal
Natural killer (NK) cells provide rapid responses to viral-infected cells and play a critical role in tumour immunosurveillance by directly inducing the death of tumour cells. Instead of acting via antigen-specific receptors, the lysis of tumour cells is mediated by alternative receptors, including NKp30.
Using computational tools, we designed a family of small organic molecules (SOMs) based on the structure of the NKp30 receptor [
5,
6]. Synthetic, stability and overall binding score considerations were used to select a subfamily of ca. 15 compounds. From these, 10 completely characterised entities were tested in an MS-based binding assay using the extracellular portion of the receptor, which led to the identification of one lead compound.
Primary cultures of human NK cells were used to probe the stimulation of NK cell responses by the lead compound. EC50 values below 0.2 µM were found in TNF-α, IFN-γ and Granzyme B release assays. Co-cultures of NK cells and the tumour cell line HCT116 were used to determine the effect of the lead compound on the cytotoxicity of NK cells. The NK cell cytotoxicity doubled upon treatment with 1.25 µM of the lead compound, as compared to control incubations, presenting an EC50 value of ca. 0.15 µM.
Our lead compound was proven to be effective in activating the cytotoxic activity of NK cells, as demonstrated by the cytokine release and the tumour cell death assays. Further work aims to derivatise our ligands with tumour-targeting molecules to increase the specificity of the NK cell cytotoxic response.
Acknowledgments
This work was supported by Fundação para a Ciência e a Tecnologia (RECI/QEQ-MED/0330/2012; SAICTPAC/0019/2015; PTDC/QUI-QAN/32242/2017; SFRH/BD/110945/2015; UID/QUI/00100/2013; UID/QUI/00100/2019) and Liga portuguesa Contra o Cancro (LPCC/NRS—Terry Fox 2015-17 grant).
2.3. Immune Responses Elicited by Photodynamic Therapy with Redaporfin
Catarina S. Lobo 1,*,
Lígia C. Gomes-da-Silva 1
and
Luís G. Arnaut 1
1
CQC, Department of Chemistry, University of Coimbra, 3004-531 Coimbra, Portugal
2
LaserLeap Technologies, 3025-307 Coimbra, Portugal
Photodynamic therapy (PDT) relies on the administration of a photosensitiser (PS) that is activated on the target tissue after irradiation with light of a specific wavelength absorbed by the PS. Redaporfin is a recently developed photosensitiser for PDT that is currently in phase 2 clinical trials (NCT02070432). A vascular protocol of redaporfin-PDT with mice bearing CT26.WT tumours not only destroys the primary tumour but also reduces the development of metastasis, thus suggesting antitumour immunity [
7].
This immune response triggered by redaporfin-PDT was further characterised and revealed a strong neutrophilia, with a systemic increase of IL-6 and an increased percentage of activated CD4+ and CD8+ T cells. At the tumour bed, it was also demonstrated that T cell tumour infiltration disappeared after PDT but reappeared with a much higher incidence one day later. Furthermore, the depletion of specific immune populations suggested that neutrophils and cytotoxic T cells play a major role in the development of the antitumour immune response elicited by redaporfin-PDT [
8].
Regarding this, it was theorised that the combination of redaporfin-PDT with an immune therapy may potentiate the efficacy of both therapies. The combination of redaporfin-PDT with immunotherapies using CTLA-4 and PD-1 was reported in three different tumour models (CT26.WT colon carcinoma, B16F10 melanoma and 4T1 mammary carcinoma models). Treatment outcomes were evaluated by survival, tumour growth kinetics and, for the carcinoma model, an observation of metastases development by bioluminescent imaging. Furthermore, changes in the expression of several immune checkpoint molecules triggered by redaporfin-PDT in vitro were also assessed.
The combination of redaporfin-PDT with CTLA-4 immunotherapy, but not with PD-1, led to a significant improvement of survival and a higher cure rate in the colon carcinoma animal model. Even though the same impact on survival was not achieved with the melanoma and breast carcinoma animal models, an interesting impact on the kinetics of metastases development was verified with the breast carcinoma model. The expression of immune checkpoint molecules was induced in tumour cells treated in vitro with redaporfin-PDT. The most notable changes were observed for CD80 and PD-L1. These results demonstrate that the combination of photodynamic therapy with immunotherapy may improve the treatment of malignant diseases that represent a challenge to immunotherapies alone and highlight that the combinatorial approaches are not universal and have to be tailored to the specificities of each clinical case.
Acknowledgments
This work was financially supported by Fundação para a Ciência e a Tecnologia (FCT) through the project Pest-OE/QUID/QUI/00313/2019 and the grants PD/BD/132524/2017 and PTDC/QEQ-MED/3521/2014.
2.4. The First Carbohydrate-Based Compounds Able to Inhibit Aβ-Induced Fyn Activation and Tau Phosphorylation: Synthesis and Biological Evaluation
Ana M. de Matos 1,*,
Maria T. Blazquez-Sanchez 1,
Cleide S. Souza 2,
Imane G. El Idrissi 3,
Nicola A. Colabufo 3,
Beat Ernst 4,
Beining Chen 2
and
Amelia P. Rauter 1,*
1
Centro de Química Estrutural, Faculdade de Ciências, Universidade de Lisboa, 1749-016 Lisboa, Portugal
2
Department of Chemistry, The University of Sheffield, Sheffield S3 7HF, UK
3
Dipartimento di Farmacia-Scienze del Farmaco, Università degli Studi di Bari “A. Moro”, 70125 Bari, Italy
4
Department of Pharmaceutical Sciences, University of Basel, CH-4056 Basel, Switzerland
Alzheimer’s disease (AD) is the most common type of dementia and is still lacking effective disease-modifying therapies to this day. This pathology is characterised by the presence of extracellular deposits of amyloid beta (Aβ) and intracellular neurofibrillary tangles containing hyperphosphorylated Tau, whose accumulation ultimately leads to synaptic dysfunction and neuronal death. Moreover, the cellular prion protein (PrPC) located in the neuronal cell surface works as a high-affinity binding partner of Aβ oligomers (Aβo). The interaction between these two players results in Fyn kinase activation with subsequent Tau hyperphosphorylation, and thus the inhibition of Aβ-induced Fyn activation is increasingly recognised as an important strategy for the treatment of Alzheimer’s disease [
9].
In this work, we present a small library of sugar-linked polyphenols with neuroprotective potential—the first carbohydrate-based compounds able to significantly inhibit Aβ-induced Fyn activation and subsequent Tau phosphorylation. Some of these molecules were also found to be Fyn kinase inhibitors. With a much more straightforward and efficient synthesis compared to other drug-like molecular entities with similar bioactivity in this therapeutic context, the most promising compounds, 1-[2,4,6-trihydroxy-3-(2,3,4,6-tetra-
O-methyl-β-D-glucopyranosyl)phenyl]ethan-1-one and 4-(β-D-glucopyranosyl) benzene-1,2-diyl dibenzoate were not cytotoxic towards neuronal cells in concentrations of up to 100 μM and 50 μM, respectively, while displaying favourable physicochemical characteristics that make them good candidates for further studies. In this communication, the synthesis and biological activity of these molecules will be explored in detail, and the potential for glucosylpolyphenols to be used as therapeutic agents against Alzheimer’s Disease will be discussed [
10].
Acknowledgments
The EU is gratefully acknowledged for having supported the project “Diagnostic and Drug Discovery Initiative for Alzheimer’s Disease”, FP7-PEOPLE-2013-IAPP, GA 612347 and Fundaçãoo para a Ciência e Tecnologia, Portugal, for supporting Centro de Química Estrutural (project UIDB/00100/2020).
2.5. Immune Checkpoint Blockade by Novel Small-Molecule Inhibitors Restores T Cell Function
Rita C. Acúrcio 1,*,
Sabina Pozzi 2,
Barbara Carreira 1,
Marta Pojo 3,
Sandra Casemiro 4,
Paula Leandro 1,
Adelaide Fernandes 1,
Andreia Barateiro 1,
Luís Costa 4,
Luís Graça 4,
Ronit Satchi-Fainaro 2,
Rita C. Guedes 1
and
Helena F. Florindo 1,*
1
Research Institute for Medicines (iMedULisboa), Faculty of Pharmacy, University of Lisbon, 1649-004 Lisbon, Portugal
2
Department of Physiology and Pharmacology, Sackler Faculty of Medicine, Tel Aviv University, 6997801 Tel Aviv, Israel
3
Instituto Português de Oncologia de Lisboa Francisco Gentil E.P.E, Unidade de Investigação em Patobiologia Molecular UIPM, Lisboa, Portugal
4
Instituto de Medicina Molecular–João Lobo Antunes, Faculty of Medicine, University of Lisbon, 1649-004 Lisboa, Portugal
5
Centro de Química Estrutural–Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal
Inhibiting the programmed cell death protein 1 (PD-1) axis has become one of the most effective therapies for diverse cancers. So far, only monoclonal antibodies (mAb) are approved as PD-1/PD-L1 inhibitors. Despite their exciting outcomes, most patients do not benefit from these antibody-based drugs, and many develop severe immune-related adverse events, precluding the full potential of the immune checkpoint blockade from being fulfilled. Here, we present the identification and validation of new PD-1/PD-L1 small-molecule inhibitors with enhanced therapeutic properties offering the possibility to circumvent the inherent challenges associated with mAb. We performed a computationally driven approach to identify PD-1/PD-L1 small-molecule inhibitor candidates. Following biochemical experiments, we have confirmed 16 in silico candidates as true inhibitors of PD-1/PD-L1 interaction. These results were subsequently corroborated in vitro. Furthermore, we assessed their impact on the T-cell function by exploiting 2D and 3D multi-cellular cancer models, using patient-derived peripheral blood mononuclear cells (PBMC) and autologous tumour cells. Finally, an in vivo study in a humanised mouse model was performed to access small-molecule anti-tumour effects. Overall, these studies showed that the newly-identified small molecules restored T-cell function by targeting the PD-1/PD-L1 co-inhibitory interaction, and this has proven to be a powerful strategy for drug discovery in the cancer immunotherapy field. The possible immune checkpoint modulation following a small molecule-based approach can revolutionise immunotherapeutic approaches in a way that can overcome some of the monoclonal antibody limitations, such as adverse events or targeting other cellular sources of PD-L1 that are critical to achieving better clinical outcomes.
Acknowledgments
This work was supported by UIDB/04138/2020, UIDP/04138/2020, PD/BD/128238/2016 (FCT-MCTES), La Caixa Health Research 2018 Call the Project proposal HR18-00589 (RSF and HF), by ENMed/0051/2016 (Israeli Ministry of Health and FCT-MCTES to HF and RSF).
2.6. Toxicokinetic/Toxicodynamic Studies of Pentedrone and Methylone: Enantioselective Approach
Bárbara Silva 1,2,*,
Carla Fernandes 2,3,
Paula Guedes de Pinho 1
and
Fernando Remião 1,*
1
UCIBIO-REQUIMTE, Laboratório de Toxicologia, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal
2
Laboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal
3
Centro Interdisciplinar de Investigação Marinha e Ambiental (CIIMAR), 4050-208 Matosinhos, Portugal
The persistent abuse of synthetic cathinones over the years leads to a huge public health concern. Since these drugs are chiral, it is important to evaluate the enantioselectivity in toxicokinetic and toxicodynamic processes [
11]. Herein, the synthetic cathinones pentedrone and methylone were the focus of this work. Initially a semi-preparative liquid chromatography (LC) method using a polysaccharide-based stationary phase was developed, to isolate both enantiomers of pentedrone and methylone. A high enantiomeric purity (superior to 97%) was obtained for all enantiomers [
12]. Then, the differentiated intestinal permeability of these cathinones was evaluated using the Caco-2 cell line. For this study, an LC method was developed and validated to quantify these drugs in the transport buffer. After the quantification of each enantiomer that passed the cell monolayer, it was possible to verify for the first time that
R-(-)-pentedrone and
S-(-)-methylone were the enantiomers most able to permeate the membranes [
13]. The third objective of this work was to evaluate the enantioselectivity of pentedrone and methylone in their neurotoxicity and also the respective underlying mechanisms using differentiated SH-SY5Y cells. The results obtained demonstrated an agreement between the enantioselectivity observed in its cytotoxicity and in oxidative stress induction, with
S-(+)-pentedrone and
R-(-)-methylone being the most cytotoxic and also the most oxidative enantiomers. Multidrug resistance-associated protein 1 (MRP1), together with glutathione (GSH), played a selective protective role against cytotoxicity induced by the
R-(-)-pentedrone. Enantioselectivity was also observed for pentedrone and methylone in its P-glycoprotein (P-gp) binding,
R-(-)-pentedrone and
S-(-)-methylone being the most transported enantiomers [
14]. Additionally, the hepatic cytotoxic and metabolic profiles of pentedrone and methylone enantiomers were assessed. The cytotoxic and metabolic profile was evaluated in 2D-HLCs and 3D-HLCs, showing that these synthetic cathinones produced cytotoxicity in the two liver models used and also an enantioselectivity in the cytotoxicity of pentedrone in the 3D model, where
R-(-)-pentedrone was the most cytotoxic enantiomer. The metabolic assay demonstrated a different profile between the enantiomers of methylone and pentedrone, with
R-(+)-methylone and
R-(-)-pentedrone being the most metabolised enantiomers.
In conclusion, this work represents an important advance related to the enantioresolution and chemical characterisation of these two synthetic cathinones. Additionally, the in vitro studies carried out in this work demonstrated, for the first time, the enantioselectivity of pentedrone and methylone in important mechanisms underlying the different toxicokinetic and toxicodynamic processes of these drugs of abuse.




{kind=link}
