
A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy †


1
Departamento de Química, Universidad de Guanajuato, Noria Alta S/N, Col. Noria Alta, 36050 Guanajuato, Mexico
2
Departamento de Química Analítica, Universidad Autónoma de Nuevo León, Facultad de Medicina, Fco. I. Madero s/n, Col. Mitras Centro, Monterrey, 64460 Nuevo León, Mexico
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry,
15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 59; https://doi.org/10.3390/ecsoc-24-08421
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:A series of six novel amide-xanthate products containing several fluorine atoms were prepared in moderate to good yields (40–92%) via an isocyanide-based multicomponent reaction (IMCR) of 5-CR by Ugi-4CR followed by an SN2 sequence in a one-pot manner. The design of molecules with fluorine atoms is of interest in medicinal chemistry and a research line of our interest. The role of fluorine atoms in biological properties is well documented, improving bioavailability, lipophilicity, and metabolic resistance in bioactive molecules.
1. Introduction
Multicomponent reactions (MCRs) are defined as reactions in which three or more starting materials react to form a product where basically all or most of the atoms contribute to the newly formed product. MCRs have many advantages over traditional multi-step sequential reactions [1]. Among the MCRs, the isocyanide-based multicomponent reactions (IMCRs) are most relevant for the synthesis of peptidomimetics. The Ugi four-component reaction (Ugi-4CR) is probably one of the most utilized IMCRs during the last decade. This reaction allows the rapid preparation of α-acylamino amides derivatives. Ugi-4CR products can exemplify a wide variety of substitution patterns and are precursors to the synthesis of peptidomimetics that have potential pharmaceutical applications [2].
The role of fluorine atoms in organic compounds to improve bioavailability, lipophilicity, and metabolic resistance in bioactive molecules is well documented [3]. Therefore, the synthesis of fluoro α-acylamino amides as a synthetic platform can be an important alternative for the synthesis of heterocycles which is of interest in medicinal chemistry.
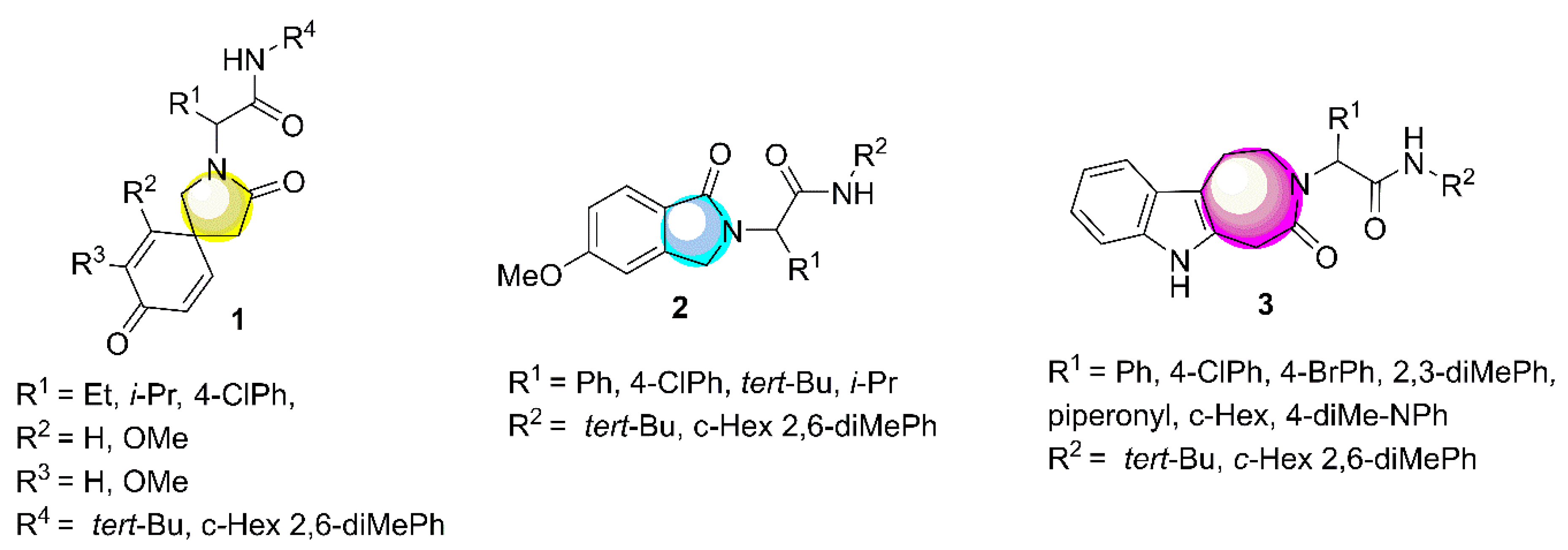
On the other hand, xanthate is an interesting functional group and has synthetic potential from radical chemistry [4]. Therefore, the one-pot incorporation of this functional group after an IMCR is an interesting strategy for the synthesis of privileged heterocyclic peptidomimetics (PHPs) via post-MCR transformation. Examples of these are azaspirodienones (1), oxindoles (2), tetrazol-azepinoindolones (3) (Figure 1) [5,6,7].
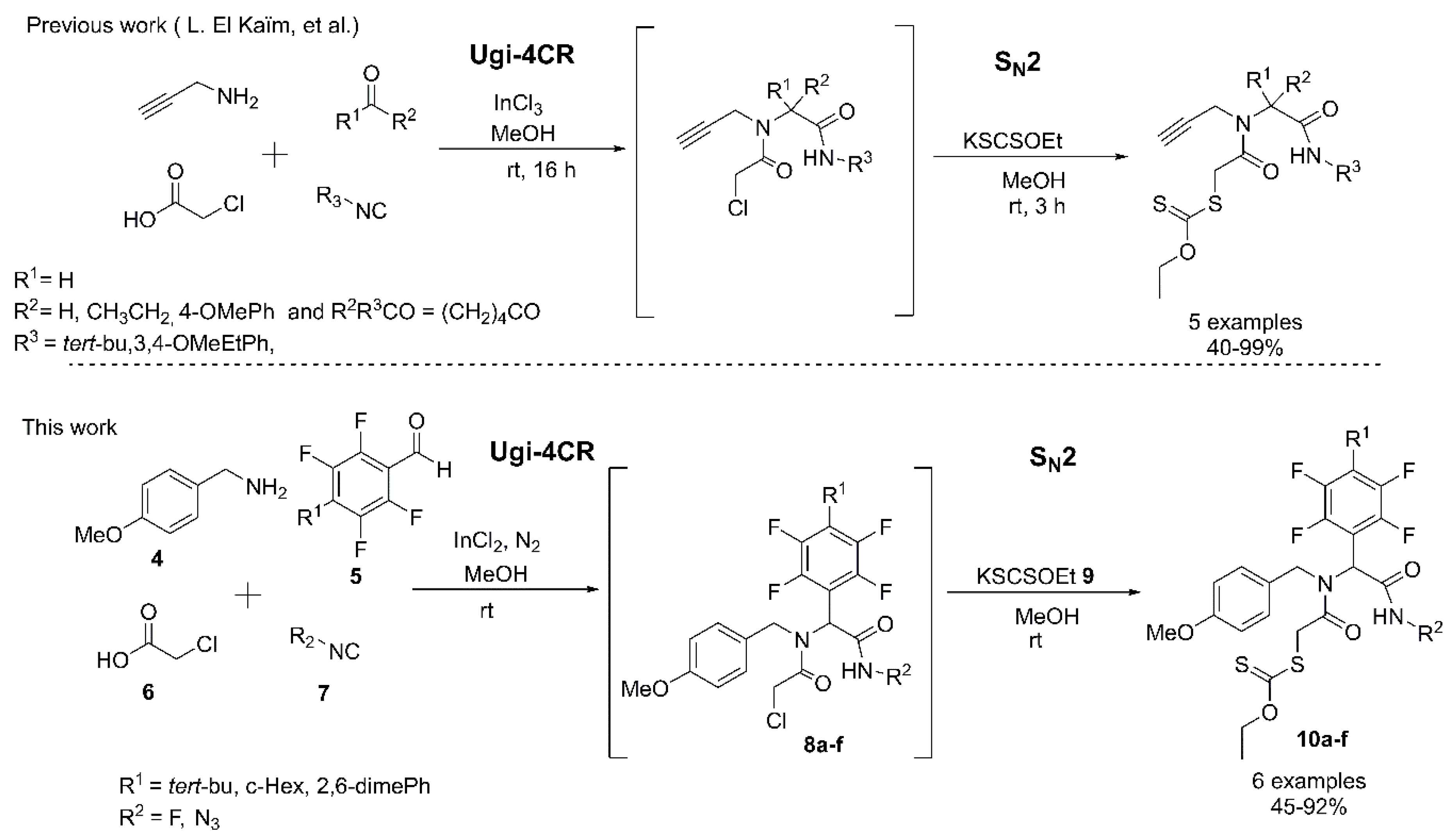
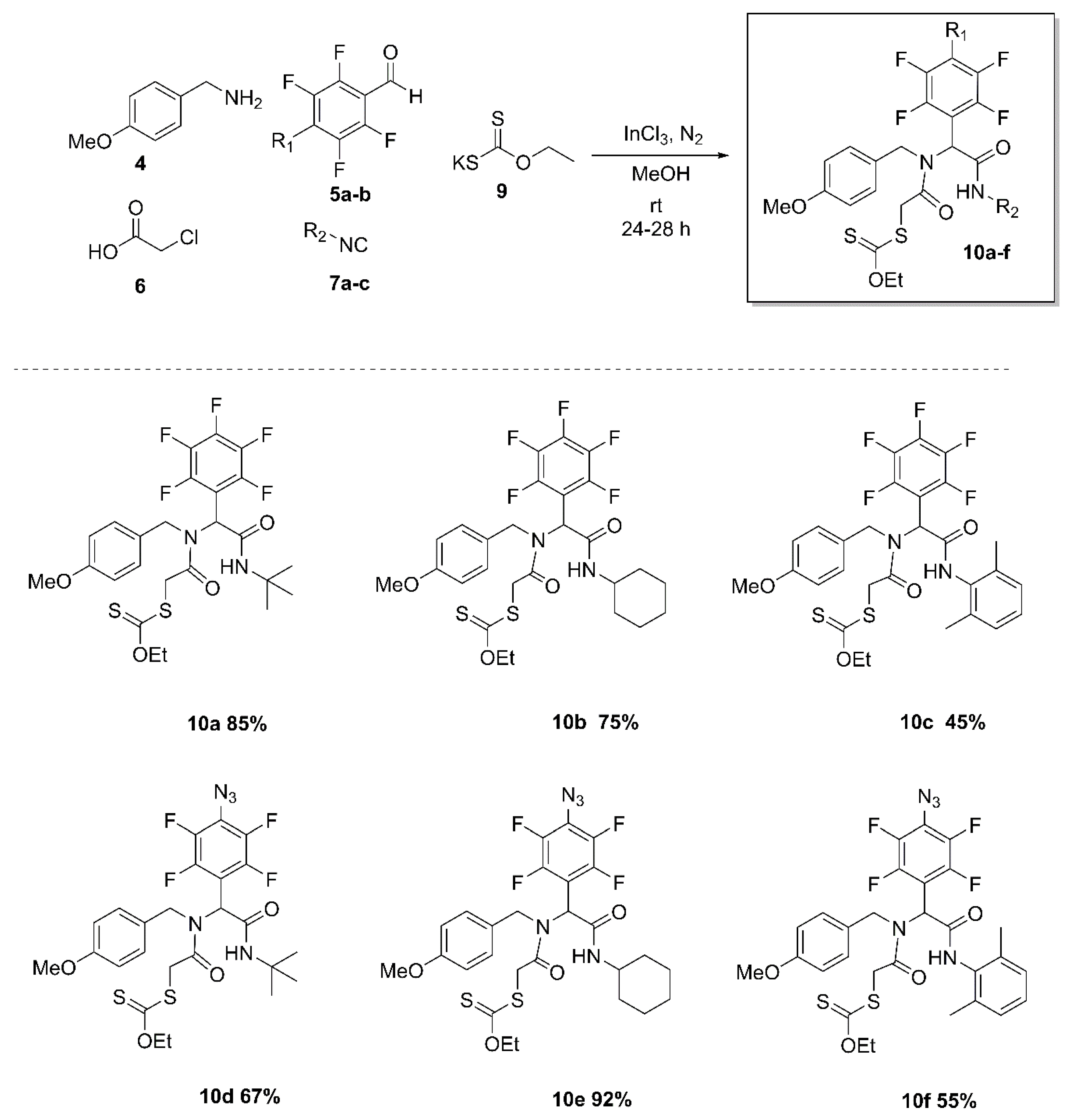
The methodology described here allows us to perform the one-pot synthesis via Ugi-4CR coupled SN2 strategy of fluoro α-acylamino amide-xanthates (10a–f). These complex xanthate products are important precursors for the synthesis of PHP products (Scheme 1).
2. Results and Discussion
In order to develop conditions for the synthesis of α-acylamino amide-xanthates, we started the optimization for the synthesis of the α-acylamino amide analogue 8d by reacting 4-methoxybenzylamine (4), 4-azido-2,3,5,6-tetrafluorobenzaldehyde (5), chloroacetic acid (6), and tert-butyl isocyanide (7) in methanol anhydrous. Initially, we performed the Ugi-4CR at room temperature, in the absence of a catalyst, the 8d product was generated in 72% after 7 days (Entry 1, Table 1). The success of the Ugi-4CR lies in the formation of the imine, which was favored by Lewis acids. When 10 mol% InCl3 was employed as catalyst, the 8d was obtained with a yield of 35% after a reaction time of 3 h (Entry 2). The yield was increased when the reaction was performed during 24 h. Then, a yield of 66% was obtained. (Entry 3). The use of microwave (MW) did not lead to the desired product and the reaction did not show a significant progress after one hour at 50 °C (Entry 4). The best reaction conditions were obtained for 20% mol of InCl3 and 1.4 equiv. carboxylic acid, after 24 h of reaction at room temperature (Entry 5).
Once the synthesis of the Ugi-4CR products was optimized, the one-pot synthesis of the α-acylamino amide-xanthates was carried out through an SN2-type reaction when adding the potassium salt of xanthic acid to form the Ugi-product xanthate 10d. The versatility of the developed methodology was examined using different aldehydes, such as 4-azido-2,3,5,6-tetrafluorobenzaldehyde (5a) and 2,3,4,5,6-pentafluorobenzaldehyde (5b), isocyanides as tert-butyl, cyclohexyl, and 2,6-dimethylphenyl (7a–c). The respective amide-xanthate products 10a–f were obtained in moderate to good yields (40–92%) (Scheme 2).
3. Experimental Section
3.1. General Information, Instrumentation, and Chemicals
1H and 13C NMR spectra were acquired on Varian Gemini spectrometers (200 MHz) and Varian Unity (300 MHz). The solvent used was deuterated chloroform (CDCl3). Chemical shifts are reported in parts per million (δ/ppm). The internal reference for 1H NMR spectra is trimethylsilane at 0.0 ppm. The internal reference for 13C NMR spectra is CDCl3 at 77.0 ppm. Coupling constants are reported in Hertz (J/Hz). Multiplicities of the signals are reported using the standard abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). NMR spectra were analyzed using the MestreNova software version 10.0.1–14719. IR spectra were acquired on a Perkin Elmer 100 spectrometer using an Attenuated Total Reflectance (ATR) method with neat compounds. The absorbance peaks are reported in reciprocal centimeters (υmax/cm−1). Reaction progress was monitored by Thin-Layer Chromatography (TLC) on precoated silica-gel 60 F254 plates and the spots were visualized under UV light at 254 or 365 nm. Mixtures of hexane with ethyl acetate (EtOAc) were used to run TLC and for measuring the retention factors (Rf). Flash column chromatography was performed using silica gel (230–400 mesh) and mixtures of hexane with EtOAc in different proportions (v/v) were used as the mobile phase. All reagents were purchased from Sigma-Aldrich and were used without further purification. Chemical names and drawings were obtained using the ChemBioDraw Ultra 13.0.2.3020 software package. The purity for all the synthesized products (up to 99%) was assessed by NMR.
3.2. Synthesis and Characterization of the Fluoro α-Acylamino Amide-Xanthates 10a–f
General procedure 1 (GP1): In a round-bottomed flask equipped with a magnetic stirring bar, 4-Methoxybenzylamine (1.4 equiv.) and InCl3 (10% mol) were added to a 0.3 M solution of p-methoxyaniline (1.0 equiv.) in anhydrous MeOH under nitrogen atmosphere at room temperature. After 60 min, chloroacetic acid (1.4 equiv.) and InCl3 (10% mol) were added. Later, tert-butyl isocyanide (1.0 equiv.) was added. The reaction mixture was stirred for 21–25 h at room temperature and then, the potassium ethyl xanthogenate salt (1.5 equiv.) was added. The reaction mixture was stirred for 3 h at room temperature. The solvent was removed until dryness. The crude was diluted in excess of AcOEt and washed with brine. The combined organic layer was dried over anhydrous Na2SO4. The solvent was removed until dryness. The crude was purified by silica-gel column chromatography to obtain the products 10a–f.
3.2.1. S-(2-((2-(tert-Butylamino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10a)
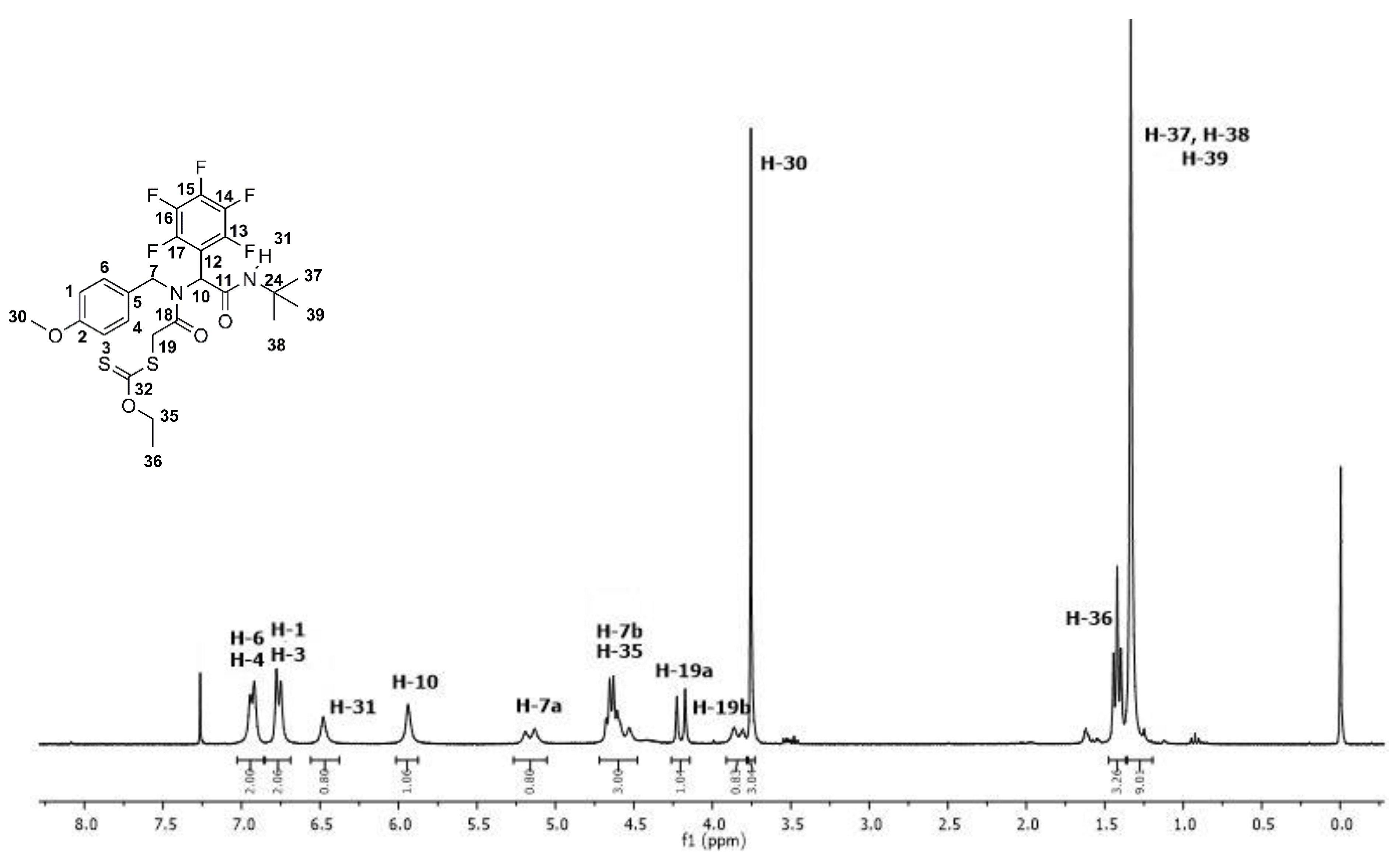
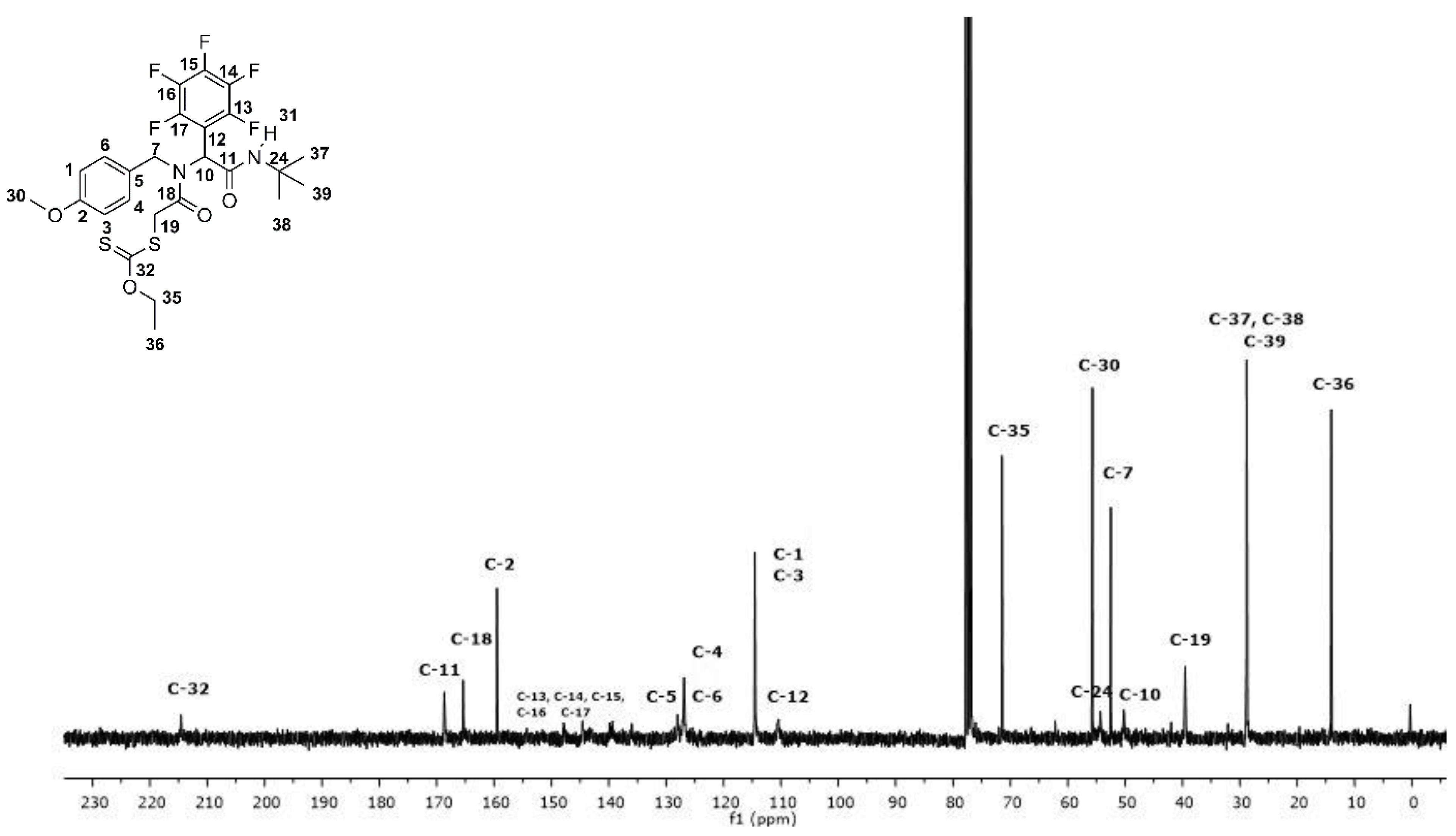
The product was obtained in 85% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.43 (hexanes–AcOEt, 8/2, v/v); mp = 130–134 °C, FT-IR (ATR) υmax/cm−1 1696, 1646 (C=O amide), 3276 (NH amide); 1H NMR (300 MHz, CDCl3, TMS): δ 6.93 (d, J = 9.0 Hz, 2H), 6.76 (d, J = 9.0 Hz, 2H), 6.48 (s, 1H), 5.94 (s, 1H), 5.16 (d, J = 18.0, 1H), 4.68 − 4.53 (m, 3H), 4.20 (d, J = 18.0 Hz, 1H), 3.83 (d, J = 18.0 Hz, 1H), 3.75 (s, 3H), 1.42 (t, J = 6.0 Hz, 3H), 1.33 (s, H, 9H); 13C NMR (75 MHz, CDCl3, TMS): δ 214.6, 168.7, 165.4, 159.5, 147.9, 144.5, 139.9, 139.4, 136.0, 128.0, 126.9, 114.5, 110.4, 71.5, 55.7, 54.3, 52.5, 50.2, 39.5, 28.8, 14.0.
3.2.2. S-(2-((2-(Cyclohexylamino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10b)
The product was obtained in 75% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.38 (hexanes–AcOEt, 8/2, v/v); mp = 148–152 °C, FT-IR (ATR) υmax/cm−1 1654, 1614 (C=O amide) 3345 (NH amide); 1H NMR (200 MHz, CDCl3, TMS): δ 6.93 (d, J = 10.0 Hz, 2H), 6.76 (d, J = 8.0 Hz, 2H), 6.55 (s, 1H), 6.26 (d, J = 8.0, 1H), 5.20 (d, J = 18.0, 1H), 4.70–4.50 (m, 3H), 4.21 (d, J = 16.0 Hz, 1H), 3.84–3.75 (m, 5H), 2.00–1.00 (m, 13H); 13C NMR (50 MHz, CDCl3, TMS): δ 215.3, 168.7, 165.4, 159.4, 148.6, 143.7, 140.2, 139.0, 135.1, 127.8, 126.8, 114.5, 110.3, 71.6, 55.6, 53.8, 50.2, 49.4, 39.3, 33.0, 25,7, 25.2, 14.0.
3.2.3. S-(2-((2-((2,6-Dimethylphenyl)amino)-2-oxo-1-(perfluorophenyl)ethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10c)
The product was obtained in 40% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.28 (hexanes–AcOEt, 8/2, v/v); mp = 166–170 °C, FT-IR (ATR) υmax/cm−1 1663, 1613 (C=O amide), 3251 (NH amide); 1H NMR (300 MHz, CDCl3, TMS): δ 7.71 (s, 1H), 7.15–7.05 (m, 3H), 6.94 (d, J = 9.0 Hz, 2H)), 6.88 (s, 1H), 6.78 (d, J = 9.0 Hz, 2H), 5.37 (d, J = 18.0, 1H), 4.70–4.53 (m, 3H), 4.29 (d, J = 18.0 Hz, 1H), 3.87–3.62 (m, 4H), 2.22 (s, 6H), 1.38 (t, J = 6.0 Hz, 3H), 1.33 (s, H, 3H); 13C NMR (75 MHz, CDCl3, TMS): δ 216.0, 169.2, 165.4, 165.1, 159.6, 136.0, 133.2, 128.6, 128.0, 127.7, 126.7, 114.7, 109.8, 71.8, 55.8, 53.7, 50.1, 39.2, 18.7, 14.0.
3.2.4. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-(tert-butylamino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10d)
The product was obtained in 67% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.38 (hexanes–AcOEt, 8/2, v/v); mp = 80–84 °C, FT-IR (ATR) υmax/cm−1 1697, 1651 (C=O amide), 2122 (N3), 3343 (NH amide); 1H NMR (300 MHz, CDCl3, TMS): δ 6.94 (d, J = 9.0 Hz, 2H)), 6.78 (d, J = 9.0 Hz, 2H), 6.47 (s, 1H), 6.92 (s, 1H), 5.15 (d, J = 18.0, 1H), 4.68–4.53 (m, 3H), 4.20 (d, J = 18.0 Hz, 1H), 3.85–3.77 (m, 4H), 1.42 (t, J = 6.0 Hz, 3H), 1.33 (s, H, 9H); 13C NMR (75 MHz, CDCl3, TMS): δ 214.4, 168.6, 165.5, 159.4, 147.7, 144.4, 142.2, 139.0, 128.2, 126.9, 120.6, 114.4, 110.7, 71.3, 55.6, 54.2, 52.2, 50.1, 39.5, 28.7, 14.0.
3.2.5. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-(cyclohexylamino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate (10e)
The product was obtained in 92% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.33 (hexanes–AcOEt, 8/2, v/v); mp = 132–136 °C, FT-IR (ATR) υmax/cm−1 1660 (C=O amide), 2123 (N3), 3378 (NH amide); 1H NMR (300 MHz, CDCl3, TMS): δ 6.94 (d, J = 9.0 Hz, 2H)), 6.78 (d, J = 9.0 Hz, 2H), 6.47 (s, 1H), 6.92 (s, 1H), 5.15 (d, J = 18.0, 1H), 4.68–4.53 (m, 3H), 4.20 (d, J = 18.0 Hz, 1H), 3.85–3.77 (m, 4H), 1.42 (t, J = 6.0 Hz, 3H), 1.33 (s, H, 9H); 13C NMR (75 MHz, CDCl3, TMS): δ 215.1, 168.7, 165.5, 159.3, 148.3, 143.0, 138.2, 137.9, 128.0, 126.9, 120.7, 114.4, 110.6, 71.5, 55.6, 53.8, 50.1, 49.4, 39.3, 33.0, 25.7, 25.2, 14.0.
3.2.6. S-(2-((1-(4-Azido-2,3,5,6-tetrafluorophenyl)-2-((2,6-dimethylphenyl)amino)-2-oxoethyl)(4-methoxybenzyl)amino)-2-oxoethyl) O-ethyl carbonodithioate e (10f)
The product was obtained in 55% as a white solid after purification by silica-gel column chromatography using a mixture of ethyl acetate with hexanes (15–20% v/v) as eluent Rf = 0.24 (hexanes–AcOEt, 8/2, v/v); mp = 136–140 °C, FT-IR (ATR) υmax/cm−1 1672, 1656 (C=O amide), 2122 (N3), 3358 (NH amide); 1H NMR (300 MHz, CDCl3, TMS): δ 6.94 (d, J = 9.0 Hz, 2H)), 6.78 (d, J = 9.0 Hz, 2H), 6.47 (s, 1H), 6.92 (s, 1H), 5.15 (d, J = 18.0, 1H), 4.68–4.53 (m, 3H), 4.20 (d, J = 18.0 Hz, 1H), 3.85–3.77 (m, 4H), 1.42 (t, J = 6.0 Hz, 3H), 1.33 (s, H, 9H); 13C NMR (75 MHz, CDCl3, TMS): δ 216.0, 169.0, 165.3, 159.5, 147.8, 143.3, 138.0, 136.0, 133.2, 128.6, 128.0, 126.8, 121.1, 114.6, 109.9, 71.8, 55.7, 53.6, 50.0, 39.2, 18.7, 14.0.
4. Conclusions
In conclusion, we have developed a one-pot strategy via the Ugi-4CR/SN2 process toward the synthesis of fluoro α-acylamino amide-xanthates. The main contributions of this work are the design and development of an MCR-based protocol towards a synthetic platform of peptidomimetic heterocycles that contain several fluorine atoms, with the aim of improving their biological properties.
Author Contributions
R.G.-M. has made a substantial, direct, and intellectual contribution to the work and M.A.R.-G. and T.R.I.-R. contributed significantly to the design and analysis of the results. All authors discussed the whole project, wrote the publication, and approved it for publication. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
RGM is grateful for financial support from DAIP-UG (154/2019, 111/2020) and CONACYT (CB-2016-285622) projects. MR-G (707974/585367) thanks CONACYT for scholarships. All authors acknowledge the Laboratorio Nacional de Caracterización de Propiedades Fisicoquímícas y Estructura Molecular (CONACYT-México, Project: 123732) for the instrumentation time provided.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Quiclet-Sire, B.; Zard, S.Z. Powerful Carbon-Carbon Bond Forming Reactions Based on a Novel Radical Exchange Process. Chem. Eur. J. 2006, 12, 6002–6016. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Montaño, R.; Ibarra-Rivera, T.; Kaïm, L.; Miranda, L. Efficient Synthesis of Azaspirodienones by Microwave-Assisted Radical Spirocyclization of Xanthate-Containing Ugi Adducts. Synthesis 2010, 8, 1285–1290. [Google Scholar] [CrossRef]
- Rentería-Gómez, A.; Islas-Jácome, A.; Jiménez-Halla, J.O.C.; Gámez-Montaño, R. Regiospecific synthesis of 1-acetamide-5-methoxy-2-oxindoles in two steps: (Ugi-SN2)/xanthate mediated free radical cyclization. Tetrahedron Lett. 2014, 55, 6567–6570. [Google Scholar] [CrossRef]
- Gordillo-Cruz, R.E.; Rentería-Gómez, A.; Islas-Jácome, A.; Cortes-García, C.J.; Díaz-Cervantes, E.; Robles, J.; Gámez-Montaño, R. Synthesis of 3-tetrazolylmethyl-azepino [4,5-b]indol-4-ones in two reaction steps: (Ugi-azide/N-acylation/SN2)/free radical cyclization and docking studies to a 5-Ht6 model. Org. Biomol. Chem. 2013, 11, 6470. [Google Scholar] [CrossRef]
- Chaturvedi, D.; Ray, S. An Efficient, One-Pot, Triton-B Catalyzed Synthesis of O-Alkyl-S-methyl Dithiocarbonates. Chem. Mon. 2006, 137, 1219–1223. [Google Scholar] [CrossRef]
- El Kaïm, L.; Grimaud, L.; Miranda, L.D.; Vieu, E.; Cano-Herrera, M.-A.; Perez-Labrada, K. New xanthate-based radical cyclization onto alkynes. Chem. Commun. 2010, 46, 2489–2491. [Google Scholar] [CrossRef] [PubMed]
- Kaïm, L.E.; Grimaud, L.; Miranda, L.D.; Vieu, E. Ugi/xanthate cyclizations as a radical route to lactam scaffolds. Tetrahedron Lett. 2006, 47, 8259–8261. [Google Scholar] [CrossRef]
Figure 1.
Some privileged heterocyclic peptidomimetics (PHPs) synthesized via post-multicomponent reactions (MCR) Table 2. and alkyl halides with a strong base as hydroxide, which is particularly problematic as it is also a substantial nucleophile, capable of hydrolyzing numerous functionalities, and it is generally restricted to alkyl substituents (see Scheme 1) [8]. The most effective methodologies for the synthesis of complex xanthates are using an IMCR followed by SN2 with potassium ethyl xanthogenate salt (see Scheme 1) [5,6,7,9].
Figure 1.
Some privileged heterocyclic peptidomimetics (PHPs) synthesized via post-multicomponent reactions (MCR) Table 2. and alkyl halides with a strong base as hydroxide, which is particularly problematic as it is also a substantial nucleophile, capable of hydrolyzing numerous functionalities, and it is generally restricted to alkyl substituents (see Scheme 1) [8]. The most effective methodologies for the synthesis of complex xanthates are using an IMCR followed by SN2 with potassium ethyl xanthogenate salt (see Scheme 1) [5,6,7,9].

Scheme 1.
Previous works and this work.

Scheme 2.
Substrate scope.

Figure 2.
1H NMR spectrum of fluoro α-acylamino amide-xanthate 10a.

Figure 3.
13C NMR spectrum of fluoro α-acylamino amide-xanthate 10a.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reaction optimizing conditions 8d.
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent c | Additive | T (°C) | Time | Yield (%) f |
| 1 a | MeOH | --- | rt | 7 days | 72 |
| 2 a | MeOH | InCl3 d | rt | 3 h | 35 |
| 3 a | MeOH | InCl3 d | rt | 24 h | 66 |
| 4 a | MeOH | InCl3 d | 50 °C MW | 1 h | nd |
| 5 b | MeOH | InCl3 e | rt | 24 h | 75 |
a Reactions performed with 1.0 equiv. 4-methoxybenzylamine (4), 1.0 equiv. of 4-azido-2,3,5,6-tetrafluorobenzaldehyde (5b), 1.0 equiv. of chloroacetic acid (6) and 1.0 equiv. of tert-butyl isocyanide (7a). b Reactions performed with 1.4 equiv. of 4-methoxybenzylamine (4), 1.0 equiv. of 4-azido-2,3,5,6-tetrafluorobenzaldehyde (5b), 1.4 equiv. of chloroacetic acid (6) and 1.0 equiv. of tert-butyl isocyanide (7a). c [1.0 M] anhydrous d 10% mol. e 20% mol f Isolated yield. rt = room temperature. nd = not determined.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rentería-Gómez, M.A.; Ibarra-Rivera, T.R.; Gámez-Montaño, R. A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy. Chem. Proc. 2021, 3, 59. https://doi.org/10.3390/ecsoc-24-08421
AMA Style
Rentería-Gómez MA, Ibarra-Rivera TR, Gámez-Montaño R. A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy. Chemistry Proceedings. 2021; 3(1):59. https://doi.org/10.3390/ecsoc-24-08421
Chicago/Turabian StyleRentería-Gómez, Manuel A., Tannya R. Ibarra-Rivera, and Rocío Gámez-Montaño. 2021. "A One-Pot Synthesis of Fluoro α-Acylamino Amide-Xanthates via an IMCR-Post Transformation Strategy" Chemistry Proceedings 3, no. 1: 59. https://doi.org/10.3390/ecsoc-24-08421