
In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results †


“Coriolan Drăgulescu” Institute of Chemistry Timisoara, 24 Mihai Viteazul Av., 300223 Timisoara, Romania
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/ .
Chem. Proc. 2021, 3(1), 25; https://doi.org/10.3390/ecsoc-24-08343
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Flavonoids, widely distributed in fruits, vegetables, and medicinal herbs, are compounds with multiple biological benefits to human health from anti-inflammatory, antioxidant, anticancer, antibacterial to antiviral activity. Coronavirus disease 2019 (COVID-19), a serious concern in the world today, is a respiratory tract disease involving moderate to severe symptoms of pneumonia, with a major incidence in older people and patients having chronic diseases. This emergency health situation led us to evaluate the possible use of natural products to prevent respiratory diseases. The present study aims to report the potential of four natural flavonoids, known to have anti-inflammatory and antiviral activity, as anti-SARS-CoV-2 through their binding on the 6YNQ protein receptor. Molecular docking study with the FRED program was chosen as an appropriate tool to analyze the interaction of natural flavonoids, quercetin, luteolin, galangin, and naringenin, with the SARS-CoV-2 main protease and to rank the conformations through a scoring function to predict their binding affinity. Overall, our preliminary results indicate the potential of the titled natural flavonoids to fight the new coronavirus, COVID-19.
1. Introduction
Coronavirus disease 2019 (COVID-19) is the most serious health concern of 2020, spreading rapidly across the continent [1]. To date, remdesivir is the only FDA-approved drug to treat COVID-19 [2,3]. Nevertheless, scientists are working hard to design new effective vaccine and “savior” protocols based on marketed anti-inflammatory and antiviral drugs or using natural resources.
SARS-CoV-2, the seventh coronavirus known to infect humans, is considered the principal causative of coronavirus disease. This virus can cause mild to severe problems at the respiratory and digestive levels. In this context, the structural inhibition of SARS-CoV-2 main protease (Mpro) at the active site can be a promising alternative for the design of selective treatment with fewer secondary effects on human health [4,5]. Some studies suggested that flavonoids can inhibit the activity of this protease [6,7,8,9]. Their inhibitory capabilities are due to the presence of numerous acceptors, donors, hydrophobic and aromatic functional units that contribute to their therapeutic action. Moreover, flavonoids are usually found in oilseeds, fruits, flowers, nuts, and vegetables, representing a vital part of the human diet, which increases the ease of treatment [10,11].
The present work aimed to assess flavonoid type compounds existing in natural resources as potential COVID-19 inhibitors, employing molecular docking and pharmacokinetic analysis. For such purpose, we have compared hydroxychloroquine [12], a drug under observational study with SARS-CoV-2 and four natural flavonoids, quercetin, luteolin, naringenin, and galangin, by analyzing their binding modes in the SARS-CoV-2 active site and pharmacokinetic profile. Galangin showed an excellent inhibitory profile against SARS-CoV-2 compared with control, hydroxychloroquine.
2. Methods
2.1. Workflow
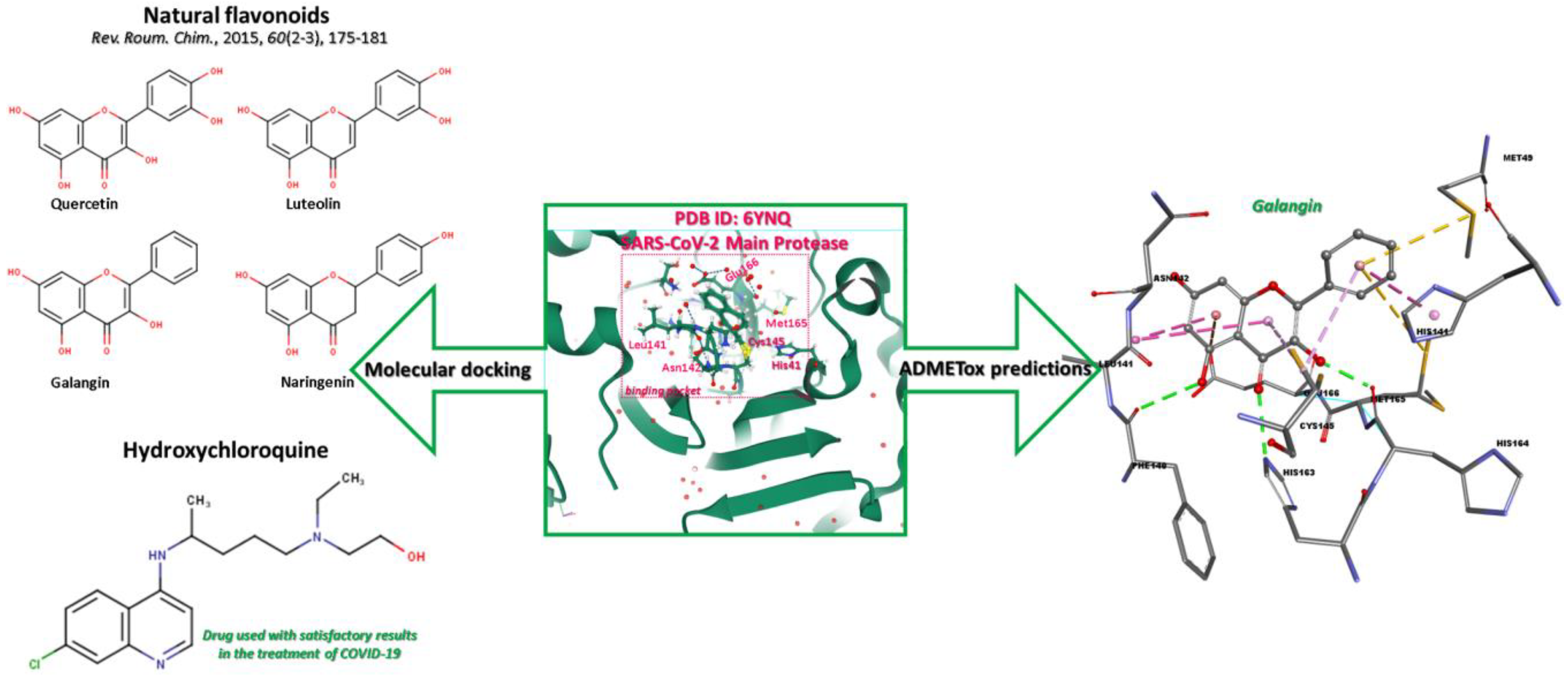
The workflow diagram (Figure 1) involves the following steps: (a) the natural flavonoids, hydroxychloroquine, and SARS-CoV-2 protein preparation, (b) molecular docking approach, and (c) the pharmacokinetic predictions
2.2. Ligands Preparation
The 3D-structure of the four natural flavonoids (quercetin, luteolin, naringenin, and galangin) and hydroxychloroquine were prepared using the Omega tool, OpenEye (OMEGA v.2.5.1.4, OpenEye Scientific Software, Santa Fe, NM, USA. www.eyesopen.com, accessed on 15 July 2020) [13,14]. The ionization states and the major microspecies of each molecule were verified at pH 7.4. These natural flavonoids were chosen to be used based on good profiles obtained from the density functional theory quantum chemical calculations (DFT) and pharmacokinetic investigations presented by Bora et al. [15]. Hydroxychloroquine was used as a standard for comparison. The generated conformations were used in docking simulations.
2.3. Protein Preparation
The X-ray structure of SARS-CoV-2 main protease (Mpro) bound to 2-methyl-1-tetralone (P6N) (PDB ID: 6YNQ) downloaded from Protein Data Bank (https://www.rcsb.org/, accessed on 15 July 2020) was prepared for docking by generating the active site box of 2400 Å3, and outer/inner contours of 1088 Å3/113 Å3 centered on the P6N ligand, using Make Receptor, OpenEye (Make Receptor, OpenEye Scientific Software, Santa Fe, NM, USA, http://www.eyesopen.com, accessed on 15 July 2020). The SARS-CoV-2 structure was selected based on the high similarity between the basic flavonoid skeleton and the co-crystallized ligands. The default parameters without imposing any constraints were applied. Water molecules of the active site that exceeded 5 Å from co-crystallized ligand were deleted. The receptor file was used further for docking experiments.
2.4. Molecular Docking
Molecular docking simulations between the titled molecules and the biological target Mpro (PDB 6YNQ) were performed with the aid of the FRED tool, OpenEye (FRED, OpenEye Scientific Software, Santa Fe, NM, USA [16,17,18]. The ligands and target proteins were treated as rigid structures during the docking simulation. Ten docking poses were retained for each ligand. To score ligand pose placement inside the active site, the Chemgauss 4 (CG4) function [16,17,18] was employed. The most likely binding conformations were retained based on the significant interactions with key residues of the active site and the highest values of the affinity score. The docking results were compared to that of the hydroxychloroquine, a repositioned drug, with satisfactory results in the COVID-19 treatment.
2.5. Prediction of Pharmacokinetic Profile
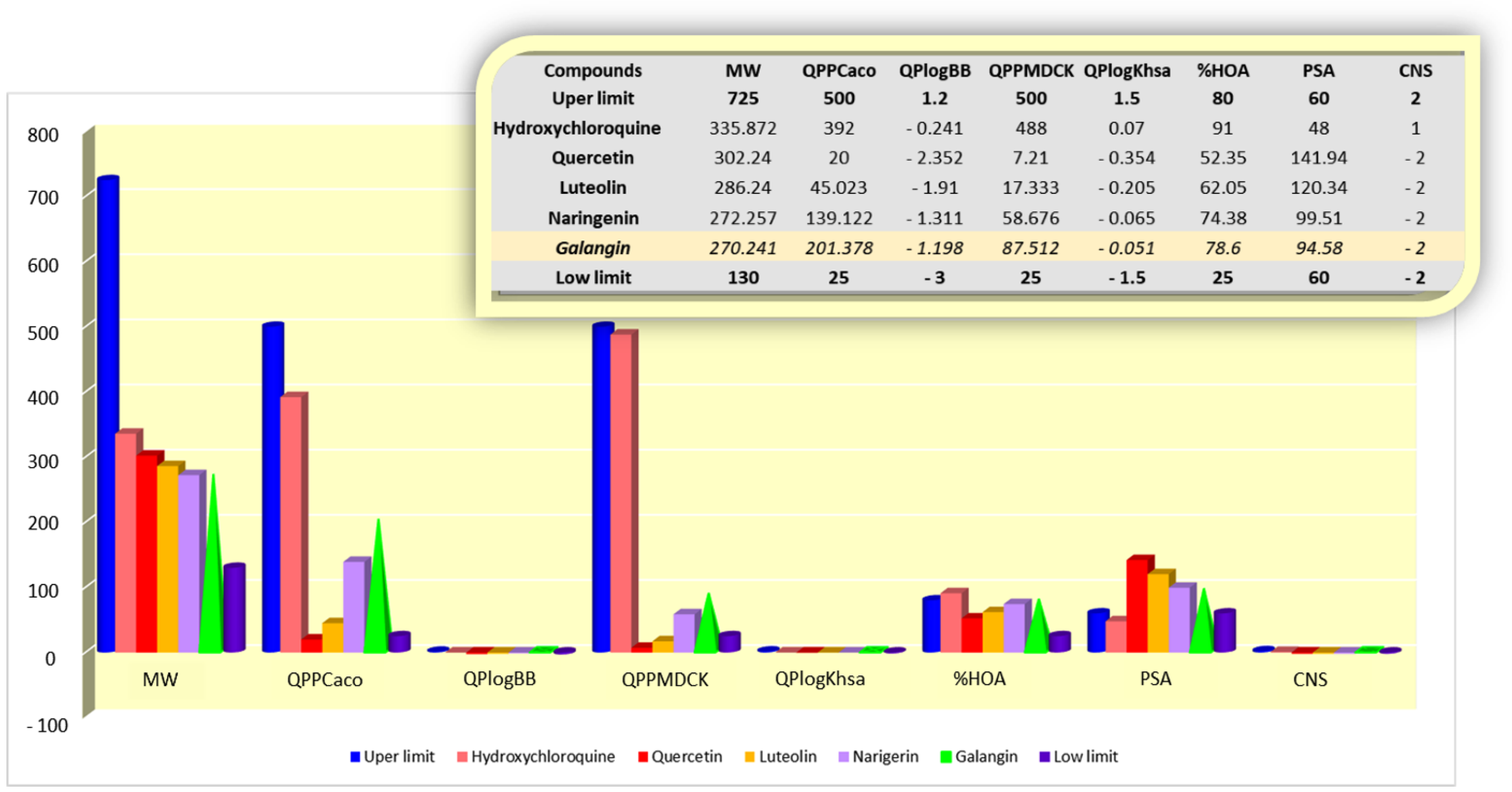
For calculation of the pharmacokinetic properties of the compounds under study with SARS-CoV-2 (quercetin, luteolin, naringenin, galangin, and hydroxychloroquine), we applied the QikProp tool, Schrodinger [19], and selecting the following special features: cell permeability (QPPCaco and QPPMDCK), binding to serum albumin (QPlogKhsa), permeability to the CNS (QPlogBB, CNS and PSA), and oral absorption (% HOA). The drug that is considered an effective candidate for COVID-9 treatment must fulfill the requirements (i) of not crossing the CNS barriers, (ii) high cellular distribution and oral absorption, and (iii) low binding to plasma protein.
The acceptable limits of the selected properties are the following:
- QPPCaco—predicted apparent Caco-2 cell permeability in nm/s: >500 (great) and <25 (poor);
- QPPMDCK—predicted apparent MDCK cell permeability in nm/s: >500 (great) and <25 (poor);
- QPlogKhsa—prediction of binding to human serum albumin: −1.5 (low) to 1.5 (high);
- QlogBB—predicted brain/blood partition coefficient: −3.0 (low) to 1.2 (easy permeability);
- CNS—predicted central nervous system activity:–2 (low permeability) to +2 (high permeability;
- PSA—van der Waals surface area of polar nitrogen and oxygen atoms: >60 does not cross the blood–brain barrier and <60 to cross the blood–brain barrier;
- % HOA—predicted human oral absorption on 0 to 100% scale: >80% (high), 25–80% (medium) and <25% (poor).
Additionally, the possible toxic action of the investigated compounds was computed using a freely accessible pkCSM platform (http://biosig.unimelb.edu.au/pkcsm/prediction, accessed on 20 September 2020). The pkCSM uses machine-learning methods to predict the pharmacokinetics and toxicity properties of small-molecules [20].
3. Results and Discussions
3.1. Molecular Docking Analysis
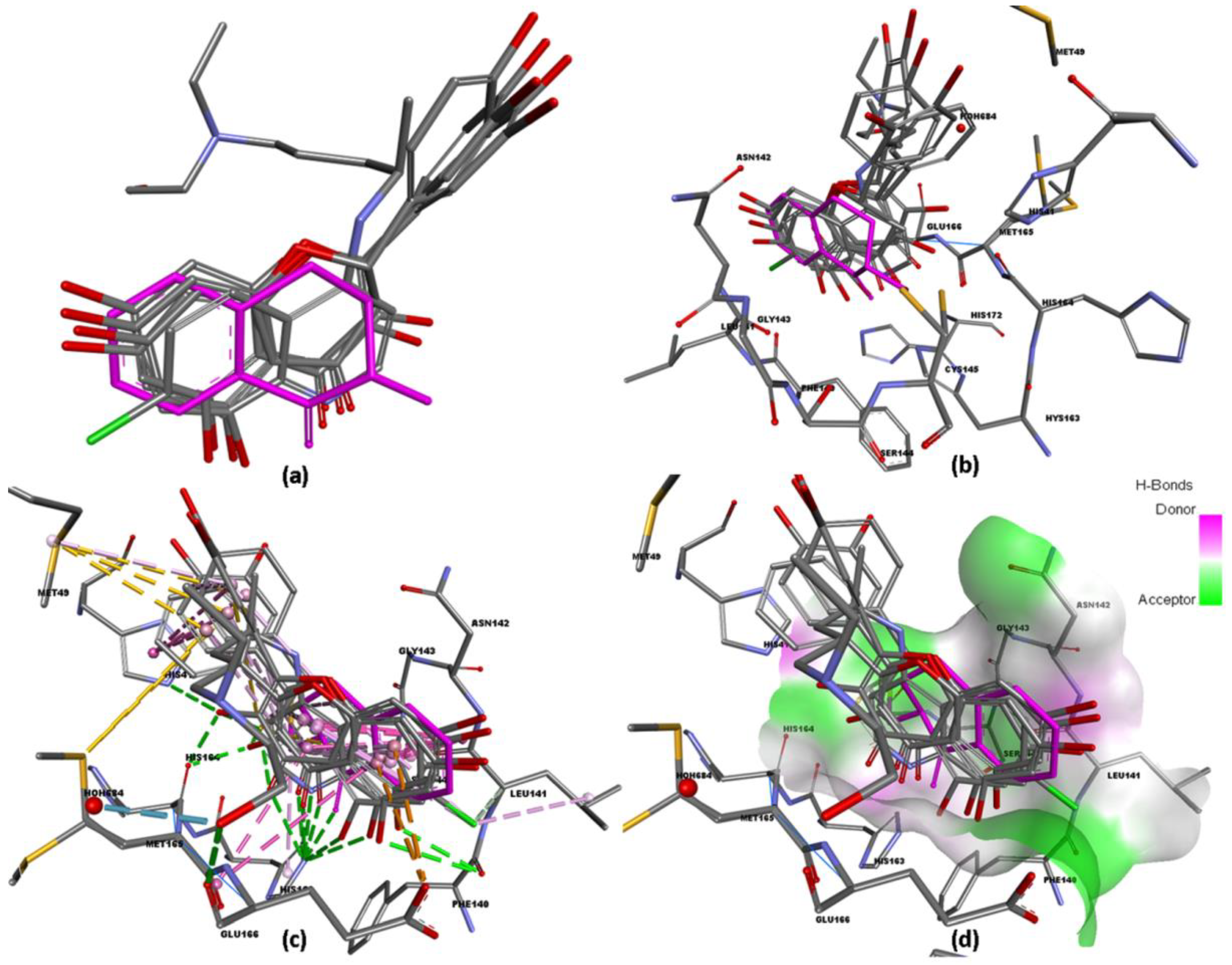
The natural flavonoid antioxidants quercetin, luteolin, naringenin and, galangin, which are usually found in various medicinal and dietary sources, comprise numerous pharmacophoric characteristics that are relevant for their application as drug-like candidates. Alignment of these antioxidant compounds over the native P6N ligand (2-Methyl-1-tetralone) and hydroxychloroquine showed significant overlaps and similarities in binding orientations and interactions (Figure 2). Therefore, such similarities have made it clear that these four natural antioxidants can act as potential inhibitors of Mpro.
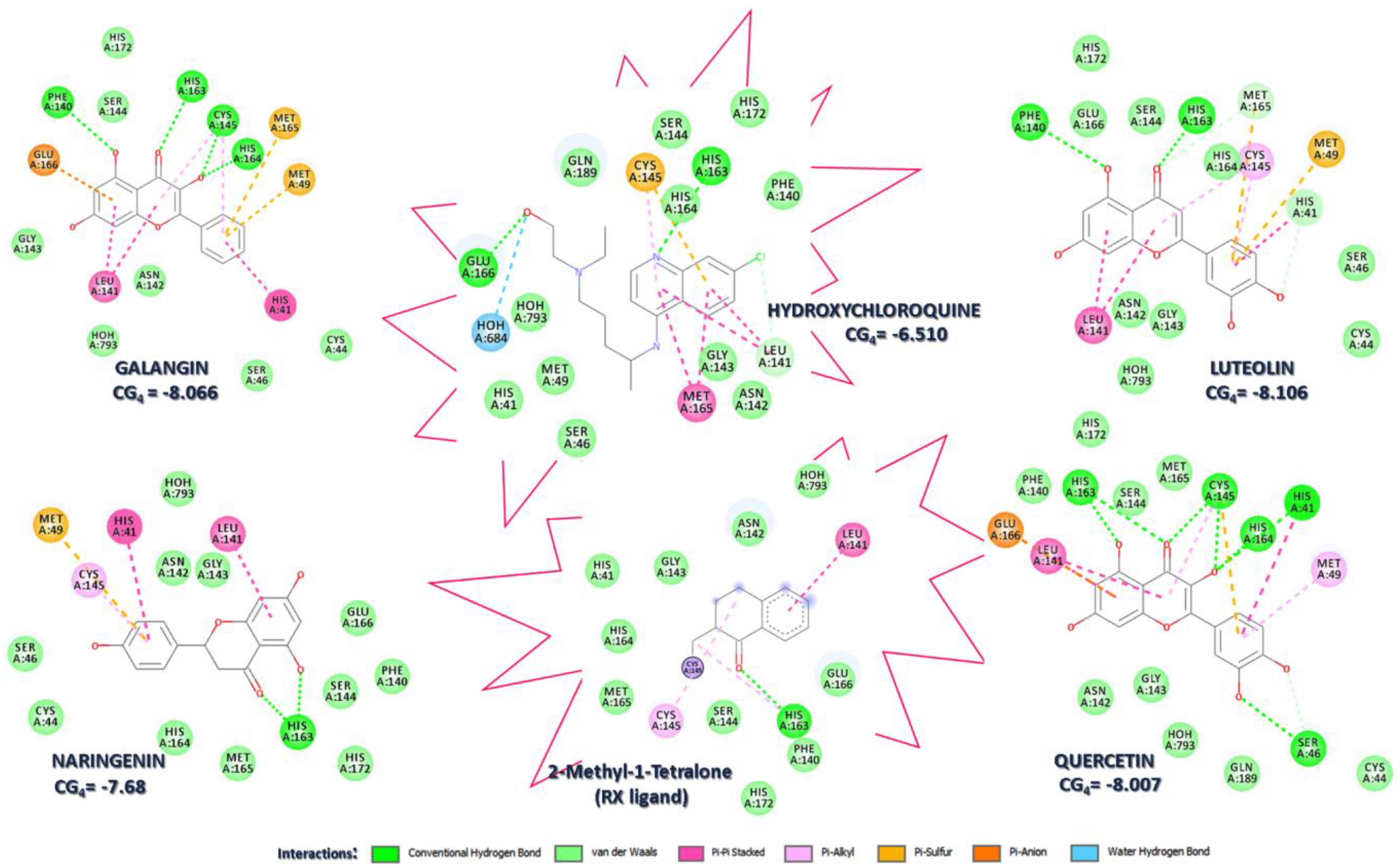
The molecular docking of natural flavonoids in the active site of SARS-CoV-2 main protease gave negative binding scores of −8.007, −8.106, −7.68, and −8.066 for quercetin, luteolin, naringenin and, galangin, respectively, which were significantly higher than the binding score of hydroxychloroquine, −6.96. The binding of these compounds into the 6NYQ active site displayed effective interactions and formation of hydrogen bonds with HIS163, HIS164, CYS145, PHE140, HIS41, and GLU166, residues (Figure 3). Moreover, interactions, such as van der Waals, п-п stacked, п-alkyl, п-sulfur, п-anion, carbon-hydrogen bonds, and water hydrogen bonds, in turn, fortified the biding orientation and contact of these ligands with the residues active site and supported our analysis to a much greater extent. The carbonyl units of all-natural flavonoids, as well as aromatic nitrogen atom of hydroxychloroquine, interact with residue HIS163. However, with the amino acid residue CYS145, only galangin and quercetin showed hydrogen bond interactions. These two flavonoids exhibited the highest number of hydrogen bond interactions involving those realized with PHE140, HIS164, and HIS41 residues. All molecules, including hydroxychloroquine, formed п-п stacked interactions between the conjugated rings and LEU141. This interaction is also observed for the native ligand, P6N. Galangin and naringenin realized additional п-п stacked interactions with HIS41. Supplementary п-sulfur and п-anion contacts with MET165, MET49, and GLU166, respectively, were observed for the natural ligands. Out of four flavonoids, galangin displayed the highest number of interactions with the largest number of amino acid residues, including (i) hydrogen bonds—4, (ii) п-п stacked—3, (iii) п-alkyl—1, (iv) п-sulfur—2 and (v) п-anion—1.
Therefore, the HIS163, HIS164, MET165, and MET49 residues demonstrated their importance in multiple interactions of different chemical natures between the ligands and the target, in addition to the HIS41 and CYS145 residues that contribute significantly to the catalytic activity of the SARS-CoV-2 main protease; we can indicate galangin as the most promising drug candidate for the treatment of COVID-19.
3.2. Pharmacokinetic and Toxicological Properties Analysis
All the investigated molecules showed results with medium to high values for human oral absorption, as well as less to median aggregation to human plasma proteins. Most of the evaluated flavonoids and hydroxychloroquine showed good cellular permeability prediction values, for both intestinal and kidney cells (QPPCaco and QPPMDCK), with the exception only of quercetin and luteolin. Excluding hydroxychloroquine, which crosses the CNS barriers, the investigated flavonoids tend not to permeate the CNS. The pkCSM predictions indicated hydroxychloroquine as exhibiting possible hepatotoxicity likely due to quinoline unit, inhibition of HERG II channels, and mutagenicity risks, demonstrating the high degree of toxicity compared with the investigated flavonoids. Excepting quercetin with possible mutagenic and tumorigenic toxicity risks, galangin, luteolin and naringenin did not show any human toxicity alerts. All the analyzed properties’ values are detailed in Figure 4.
Shortly, analyzing by comparison with the predicted pharmacokinetic profile of the hydroxychloroquine, it can be concluded that the profile obtained for galangin (Figure 4) is the most satisfactory and appropriate to be further evaluated as a clinical alternative against this pandemic disease.
Our hypothesis of SARS-CoV-2 potential inhibition with natural compounds of flavonoid type seems to be possible and requires extensive further research to design an effective natural treatment against COVID-19.
4. Conclusions
We have carried out molecular docking and pharmacokinetic studies of four natural flavonoids and hydroxychloroquine with SARS-CoV-2 main protease receptor. The docking results (binding energy and key interactions) and pharmacokinetic profiles were compared with hydroxychloroquine, a repurposed drug with potential benefits against COVID-19 [12,21,22]. Our results revealed that all flavonoids exhibited higher docking scores than hydroxychloroquine against the SARS-CoV-2 protease. One out of four flavonoids, galangin, showed pharmacological properties similar to hydroxychloroquine, with particular improvement in its potential of not permeates the CNS. Therefore, based on the promising docking and pharmacokinetic outcomes and the medicinal relevance of galangin, we suggest being further evaluated as a possible repurposed drug to combat COVID-19.
Author Contributions
A.B. and L.C. performed computational studies. A.B. edited the manuscript; L.P. evaluated some data. All authors discussed the outcomes and commented on the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors thank ChemAxon Ltd., OpenEye Ltd., and BIOVIA software Inc. (Discovery Studio Visualizer) for providing the academic license. The authors thank Schrödinger Inc. for providing an academic trial license to complete the calculations for this paper. Project No. 1.2 of the “Coriolan Dragulescu” Institute of Chemistry, Timisoara, financially supported the current approach.
Conflicts of Interest
The authors indicate no potential conflict of interest.
References
- Acter, T.; Uddin, N.; Das, J.; Akhter, A.; Choudhury, T.R.; Kim, S. Evolution of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-Cov-2) as Coronavirus Disease 2019 (COVID-19) pandemic: A global health emergency. Sci. Total Environ. 2020, 730, 138996. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Treatment for COVID-19. 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 6 October 2020).
- Guarner, J. Three emerging coronaviruses in two decades: The story of SARS, MERS, and now COVID-19. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Hage-Melim, L.I.D.S.; Federico, L.B.; de Oliveira, N.K.S.; Francisco, V.C.C.; Correia, L.C.; de Lima, H.B.; Gomes, S.Q.; Barcelos, M.P.; Francischini, I.A.G.; da Silva, C.H.T.P. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020, 256, 117963. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of natural products as potential inhibitors of COVID-19 main protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef]
- Pandey, A.K.; Verma, S. An in-silico evaluation of dietary components for structural inhibition of SARS-Cov-2 main protease. J. Biomol. Struct. Dyn. 2020, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.-E.; Lowe, H.; Toyang, N. Potential of flavonoid-inspired phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef]
- Russo, M.; Moccia, S.; Spagnuolo, C.; Tedesco, I.; Russo, G.L. Roles of flavonoids against coronavirus infection. Chem. Biol. Interact. 2020, 328, 109211. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed]
- D’Arminio Monforte, A.; Tavelli, A.; Bai, F.; Marchetti, G.; Cozzi-Lepri, A. Effectiveness of hydroxychloroquine in COVID-19 disease: A done and dusted deal? Int. J. Infect. Dis. 2020, 99, 75–76. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge structural database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Bora, A.; Avram, S.; Crisan, L.; Halip, L.; Curpan, R.; Pacureanu, L.; Ciucanu, I. Theoretical investigations of quercetin and its analogues as anticancer agents. Rev. Roum. Chim. 2015, 60, 175–181. [Google Scholar]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. QikProp 4.4; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Ingoglia, G.; Ippolito, M.; Giarratano, A.; Einav, S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020, 57, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Arshad, S.; Kilgore, P.; Chaudhry, Z.S.; Jacobsen, G.; Wang, D.D.; Huitsing, K.; Brar, I.; Alangaden, G.J.; Ramesh, M.S.; McKinnon, J.E.; et al. Henry Ford COVID-19 Task Force. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalised with COVID-19. Int. J. Infect. Dis. 2020, 97, 396–403. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Computational workflow scheme.

Figure 2.
Different perspectives of the docked protein–ligand complex. (a) The hydroxychloroquine, flavonoids, and co-crystallized ligand structures superposition (b) the binding orientation of the structures of the ligands in the 6YNQ active site (c) the significant hydrogen-bond and hydrophobic interactions established by the ligands with the key residues of the 6NYQ active site (d) the donor and acceptor surfaces around ligands.
Figure 2.
Different perspectives of the docked protein–ligand complex. (a) The hydroxychloroquine, flavonoids, and co-crystallized ligand structures superposition (b) the binding orientation of the structures of the ligands in the 6YNQ active site (c) the significant hydrogen-bond and hydrophobic interactions established by the ligands with the key residues of the 6NYQ active site (d) the donor and acceptor surfaces around ligands.

Figure 3.
Docking analysis. 2D representation of key interactions between hydroxychloroquine, natural flavonoids, co-crystallized P6N ligand, and the 6YNQ-binding site and the corresponding CG4 docking score.
Figure 3.
Docking analysis. 2D representation of key interactions between hydroxychloroquine, natural flavonoids, co-crystallized P6N ligand, and the 6YNQ-binding site and the corresponding CG4 docking score.

Figure 4.
Numerical values and graphical representation of the QikProp properties computed for hydroxychloroquine and the four natural flavonoids.
Figure 4.
Numerical values and graphical representation of the QikProp properties computed for hydroxychloroquine and the four natural flavonoids.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bora, A.; Pacureanu, L.; Crisan, L. In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results. Chem. Proc. 2021, 3, 25. https://doi.org/10.3390/ecsoc-24-08343
AMA Style
Bora A, Pacureanu L, Crisan L. In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results. Chemistry Proceedings. 2021; 3(1):25. https://doi.org/10.3390/ecsoc-24-08343
Chicago/Turabian StyleBora, Alina, Liliana Pacureanu, and Luminita Crisan. 2021. "In Silico Study of Some Natural Flavonoids as Potential Agents against COVID-19: Preliminary Results" Chemistry Proceedings 3, no. 1: 25. https://doi.org/10.3390/ecsoc-24-08343