
A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells †

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. General Procedure for the Synthesis of Analogs with Cyclopentanone and Cyclohexanone Cores
2.3. General Procedure for the Synthesis of Analogs with 4-Piperidone Cores
2.4. Computational Studies
2.4.1. Dataset
2.4.2. Geometry Optimization
2.4.3. 2D-QSAR Methodology
2.4.4. 3D-QSAR Methodology
- Quenched Molecular Dynamics and Alignment
- CoMFA Model
3. Results
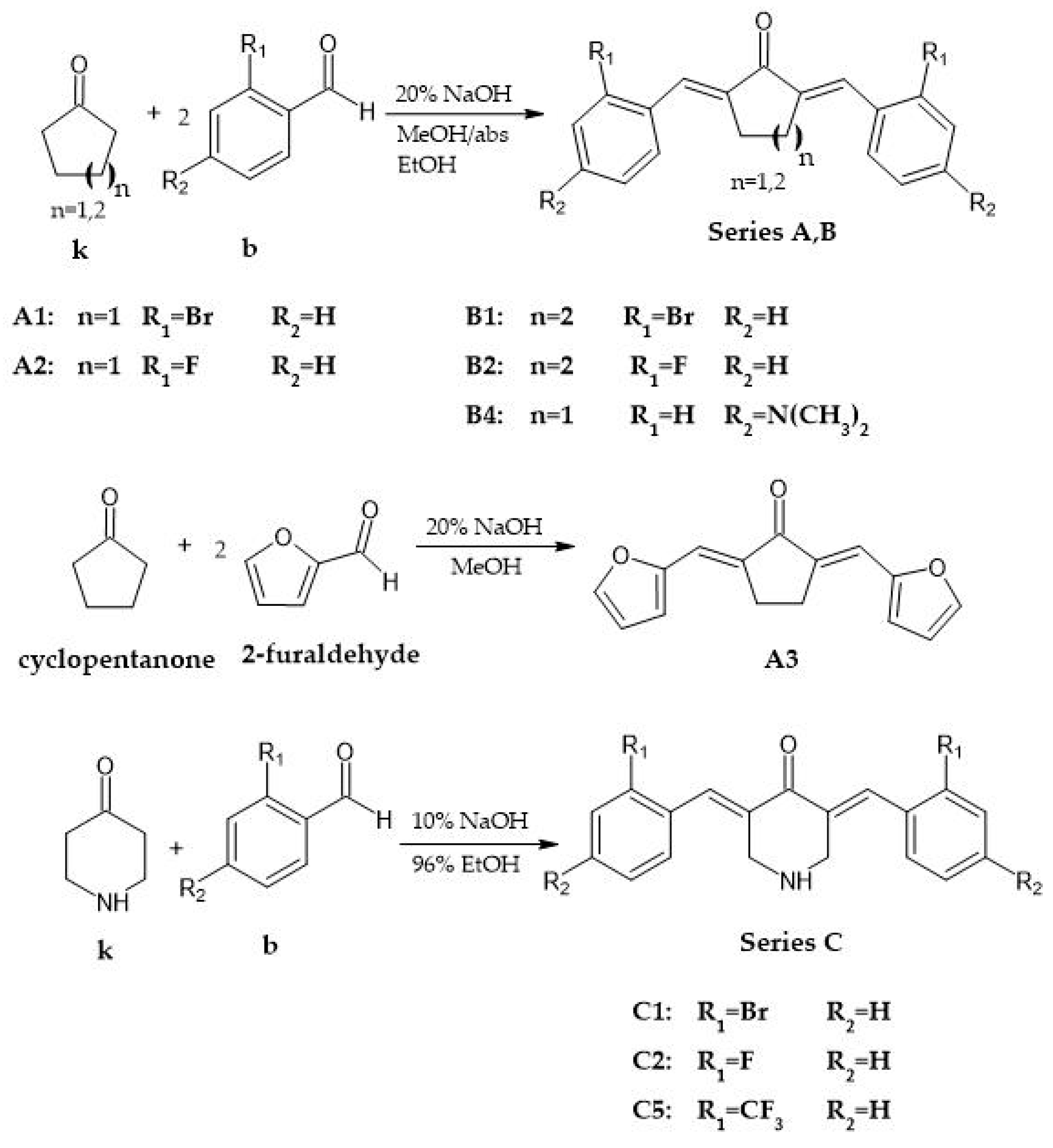
3.1. Synthesis
3.2. 2D-QSAR
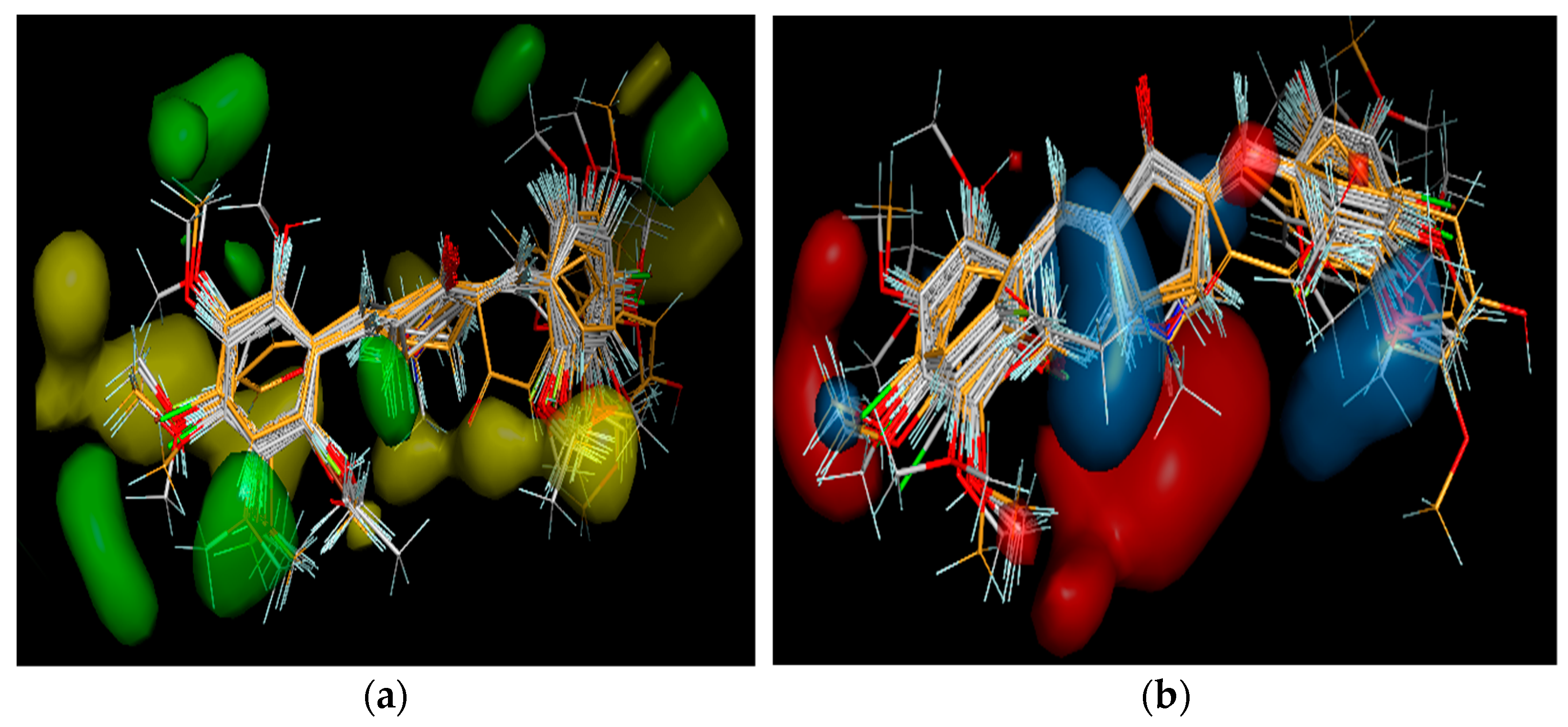
3.3. 3D-QSAR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 15 August 2021).
- Robles-Escajeda, E.; Das, U.; Ortega, N.M.; Parra, K.; Francia, G.; Dimmock, J.R.; Varela-Ramirez, A.; Aguilera, R.J. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell. Oncol. 2016, 39, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-D.; Liu, X.-E.; Huang, D.-S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol. Med. Rep. 2012, 6, 1267–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraju, G.P.; Aliya, S.; Zafar, S.F.; Basha, R.; Diaz, R.; El-Rayes, B.F. The impact of curcumin on breast cancer. Integr. Biol. 2012, 4, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, J.; Cui, R.; Lin, J.; Ding, X. Curcumin in Treating Breast Cancer: A Review. J. Lab. Autom. 2016, 21, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P.; Maurici, A.; Inguglia, L.; Marchetti, P.; Rizzi, M.; Baruchello, R.; Simoni, D.; D’Alessandro, N. Curcumin as a Possible Lead Compound against Hormone-Independent, Multidrug-Resistant Breast Cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xu, Y.; Meng, L.; Huang, L.; Sun, H. Curcumin inhibits proliferation and promotes apoptosis of breast cancer cells. Exp. Ther. Med. 2018, 16, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, M.; Dai, E.; Luo, Y. Molecular targets of curcumin in breast cancer (Review). Mol. Med. Rep. 2019, 19, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mock, C.D.; Jordan, B.C.; Selvam, C. Recent advances of curcumin and its analogues in breast cancer prevention and treatment. RSC Adv. 2015, 5, 75575–75588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the Heart from the Curcumin Molecule: Monocarbonyl Curcumin Mimics (MACs). Molecules 2014, 20, 249–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamrus, S.N.H.; Akhtar, M.N.; Yeap, S.K.; Quah, C.K.; Loh, W.-S.; Alitheen, N.B.; Zareen, S.; Tajuddin, S.N.; Hussin, Y.; Shah, S.A.A. Design, synthesis and cytotoxic effects of curcuminoids on HeLa, K562, MCF-7 and MDA-MB-231 cancer cell lines. Chem. Central J. 2018, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorganic Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, R.; Korabecny, J.; Malinak, D.; Honegr, J.; Musilek, K.; Kuca, K. Ligand-based 3D QSAR analysis of reactivation potency of mono- and bis-pyridinium aldoximes toward VX-inhibited rat acetylcholinesterase. J. Mol. Graph. Model. 2015, 56, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chu, Y.; Jiang, N.; Yang, J.; Li, F. The Three Dimensional Quantitative Structure Activity Relationships (3D-QSAR) and Docking Studies of Curcumin Derivatives as Androgen Receptor Antagonists. Int. J. Mol. Sci. 2012, 13, 6138–6155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sippl, W. 3D-QSAR—Applications, Recent Advances, and Limitations. In Recent Advances in QSAR Studies; Springer: Dordrecht, The Netherlands, 2009; pp. 103–125. [Google Scholar] [CrossRef]
- Xue, C.; Cui, S.; Liu, M.; Hu, Z.; Fan, B. 3D QSAR studies on antimalarial alkoxylated and hydroxylated chalcones by CoMFA and CoMSIA. Eur. J. Med. Chem. 2004, 39, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2010, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Papa, E.; Doucet, J.P.; Doucet-Panaye, A. Computational approaches for the prediction of the selective uptake of magnetofluorescent nanoparticles into human cells. RSC Adv. 2016, 6, 68806–68818. [Google Scholar] [CrossRef]
- Achutha, A.S.; Pushpa, V.L.; Suchitra, S. Theoretical Insights into the Anti-SARS-CoV-2 Activity of Chloroquine and Its Analogs and In Silico Screening of Main Protease Inhibitors. J. Proteome Res. 2020, 19, 4706–4717. [Google Scholar] [CrossRef] [PubMed]
- Tadayon, M.; Garkani-Nejad, Z. In silico study combining QSAR, docking and molecular dynamics simulation on 2,4-disubstituted pyridopyrimidine derivatives. J. Recept. Signal Transduct. 2019, 39, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, C.; Raghuraman, A.; Kier, L.B.; Desai, U.R. Application of Molecular Connectivity and Electro-Topological Indices in Quantitative Structure-Activity Analysis of Pyrazole Derivatives as Inhibitors of Factor Xa and Thrombin. Chem. Biodivers. 2008, 5, 2609–2620. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Statistical Parameters | Values |
|---|---|
| R | 0.881 |
| R2 | 0.775 |
| Adjusted R2 | 0.700 |
| SE | 0.267 |
| F | 10.336 |
| Significance F | 1.4 × 10−5 |
| R2 pred | 0.7827 |
| Statistical Parameters | Values |
|---|---|
| R | 0.979 |
| R2 FFDSEL | 0.959 |
| Q2loo FFDSEL | 0.401 |
| SDEP | 0.371 |
| R2 pred | 0.907 |
| SDEP pred | 0.167 |
| Analog | Predicted pIC50 Values (µmol/L) |
|---|---|
| (3E,5E)-3,5-bis(2-fluorobenzylidene)-4-piperidone | 5.46 |
| (3E, 5E)-3,5-bis(2-bromobenzylidene)-4-piperidone | 5.19 |
| (2E, 6E)-2,6-bis(2-fluorobenzylidene)cyclohexanone | 5.15 |
| (2E, 6E)-2,6-bis(2-bromobenzylidene)cyclohexanone | 5.14 |
| (3E, 5E)-3,5-bis(2-trifluoromethylbenzylidene)-4-piperidone | 5.13 |
| (2E,5E)-2,5-Bis(2-furylmethylene)cyclopentanone | 5.07 |
| (2E, 6E)-2,6-bis(4-dimethylaminobenzylidene)cyclohexanone | 4.54 |
| (2E, 6E)-2,6-bis(2-fluorobenzylidene)cyclopentanone | 4.36 |
| (2E, 6E)-2,6-bis(2-bromobenzylidene)cyclopentanone | 4.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorovska, I.; Dragarska, K.; Bogdanov, J. A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells. Chem. Proc. 2022, 12, 5. https://doi.org/10.3390/ecsoc-26-13572
Todorovska I, Dragarska K, Bogdanov J. A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells. Chemistry Proceedings. 2022; 12(1):5. https://doi.org/10.3390/ecsoc-26-13572
Chicago/Turabian StyleTodorovska, Ivana, Katerina Dragarska, and Jane Bogdanov. 2022. "A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells" Chemistry Proceedings 12, no. 1: 5. https://doi.org/10.3390/ecsoc-26-13572