
“TYC” Reaction between Alkynes and Catechol-Thiol Derivatives Prompted by Metal Nanocatalysis: Mechanism Study by DFT Calculation †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Computational Methods
2.2. Experimental Methods
Preparation of FeNPs/TiO2 catalyst
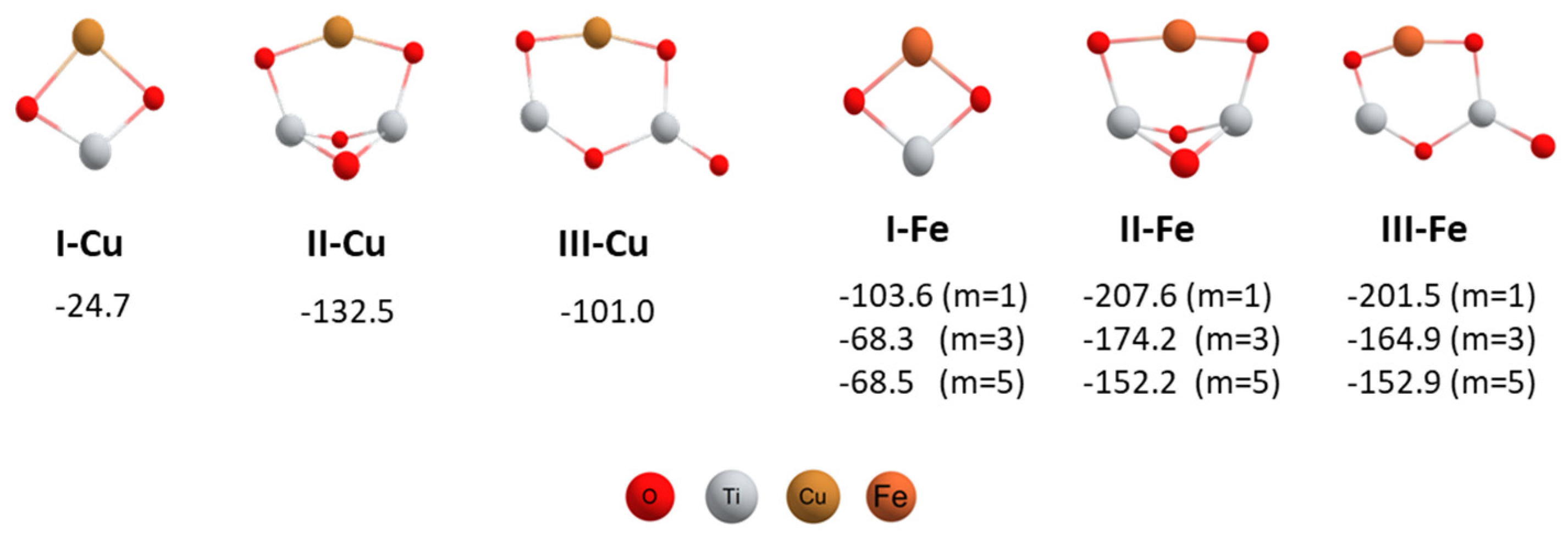
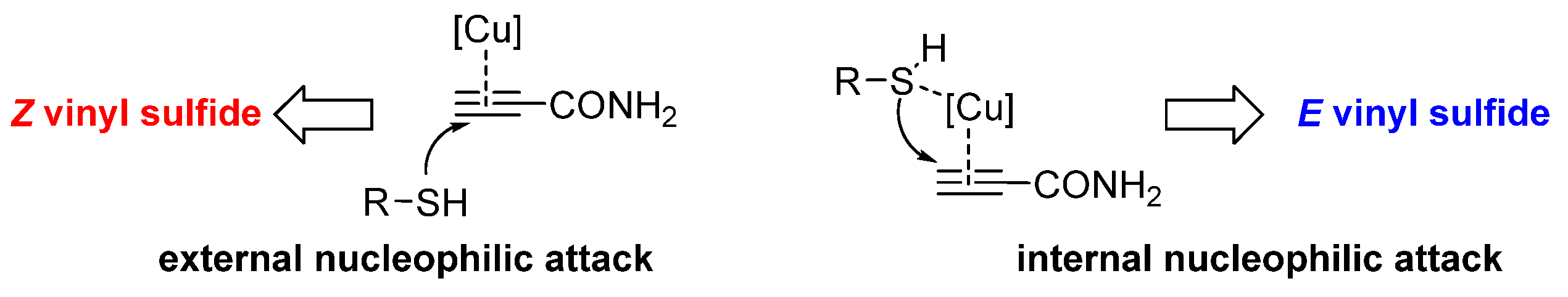
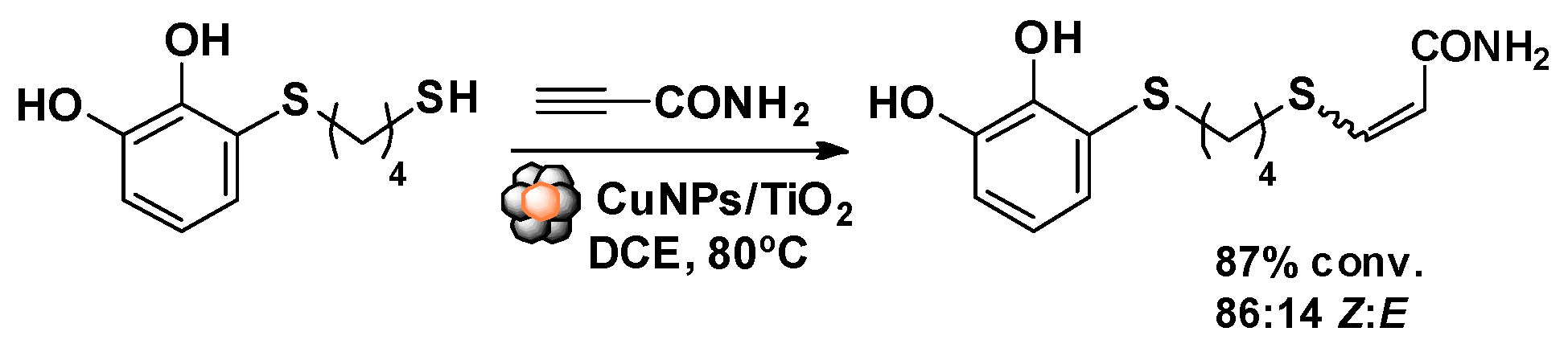
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doroszuk, J.; Musiejuk, M.; Ponikiewski, Ł.; Witt, D. Convenient and Efficient Diastereoselective Preparation of Functionalized Z-Alkenyl Sulfides. Eur. J. Org. Chem. 2018, 2018, 6333–6337. [Google Scholar] [CrossRef]
- Riesco-Domínguez, A.; van de Wiel, J.; Hamlin, T.A.; van Beek, B.; Lindell, S.D.; Blanco-Ania, D.; Bickelhaupt, F.M.; Rutjes, F.P.J.T. Trifluoromethyl Vinyl Sulfide: A Building Block for the Synthesis of CF3S-Containing Isoxazolidines. J. Org. Chem. 2018, 83, 1779. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, M.; Pacifico, S.; Chambery, A.; Marra, A.; Dondoni, A. Synthesis of S-glycosyl amino acids and S-glycopeptides via photoinduced click thiol–ene coupling. J. Org. Chem. 2010, 75, 4644. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabapathy, S.; Sudalai, A.; Benicewicz, B.C. Montmorillonite K 10-catalyzed regioselective addition of thiols and thiobenzoic acids onto olefins: An efficient synthesis of dithiocarboxylic esters. Tetrahedron Lett. 2001, 42, 3791. [Google Scholar] [CrossRef]
- Kondoh, K.A.; Takami, H.; Yorimitsu, K.; Oshima, J. Stereoselective Hydrothiolation of Alkynes Catalyzed by Cesium Base: Facile Access to (Z)-1-Alkenyl Sulfides. Org. Chem. 2005, 70, 6468. [Google Scholar] [CrossRef] [PubMed]
- Nador, F.; Mancebo-Aracil, J.; Zanotto, D.; Ruiz-Molina, D.; Radivoy, G. Thiol-yne click reaction: An interesting way to derive thiol-provided catechols. RSC Adv. 2021, 11, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e132. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L.J. Effect of the damping function in dispersion corrected density functional theory. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.J. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. Phys. Chem. A 1998, 102, 1995. [Google Scholar] [CrossRef]
- Capurso, M.; Radivoy, G.; Nador, F.; Dorn, V. Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chem. Proc. 2021, 3, 120. [Google Scholar]
- Li, S.; Wang, J.; Jacobson, P.; Gong, X.; Selloni, A.; Diebold, U. Correlation between Bonding Geometry and Band Gap States at Organic−Inorganic Interfaces: Catechol on Rutile TiO2(110). J. Am. Chem. Soc. 2009, 131, 980–984. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capurso, M.; Radivoy, G.; Nador, F.; Dorn, V. “TYC” Reaction between Alkynes and Catechol-Thiol Derivatives Prompted by Metal Nanocatalysis: Mechanism Study by DFT Calculation. Chem. Proc. 2022, 12, 4. https://doi.org/10.3390/ecsoc-26-13588
Capurso M, Radivoy G, Nador F, Dorn V. “TYC” Reaction between Alkynes and Catechol-Thiol Derivatives Prompted by Metal Nanocatalysis: Mechanism Study by DFT Calculation. Chemistry Proceedings. 2022; 12(1):4. https://doi.org/10.3390/ecsoc-26-13588
Chicago/Turabian StyleCapurso, Matías, Gabriel Radivoy, Fabiana Nador, and Viviana Dorn. 2022. "“TYC” Reaction between Alkynes and Catechol-Thiol Derivatives Prompted by Metal Nanocatalysis: Mechanism Study by DFT Calculation" Chemistry Proceedings 12, no. 1: 4. https://doi.org/10.3390/ecsoc-26-13588