
Multicenter, Open Label, Randomized Controlled Superiority Trial for Availability to Reduce Nocturnal Urination Frequency: Study Protocol for a TOP-STAR Study
 , , , , , and
, , , , , and 
Abstract
:1. Introduction
1.1. Background and Rationale
1.2. Objectives
1.2.1. Primary Objectives
1.2.2. Secondary Objectives
- Frequency of urination during the day
- Change in ratio of urinary volume at night to 24 h
- Change in urine volume at night
- Change in urine volume during the day
- Change in total urinary sodium excretion and other urinalysis
- Change in the results of blood tests
- Change in home blood pressure at night
- Change in body composition test
- Change in questionnaire score (The Diabetes Treatment Satisfaction Questionnaire, status version, DTSQs score; Diabetes Diet-Related Quality of Life Revised, DDRQOL-R; Brief-type Self-administered Diet History Questionnaire, BDHQ; Core Lower Urinary Tract Symptoms score, CLSS)
- Incidence of adverse events and diseases
2. Methods
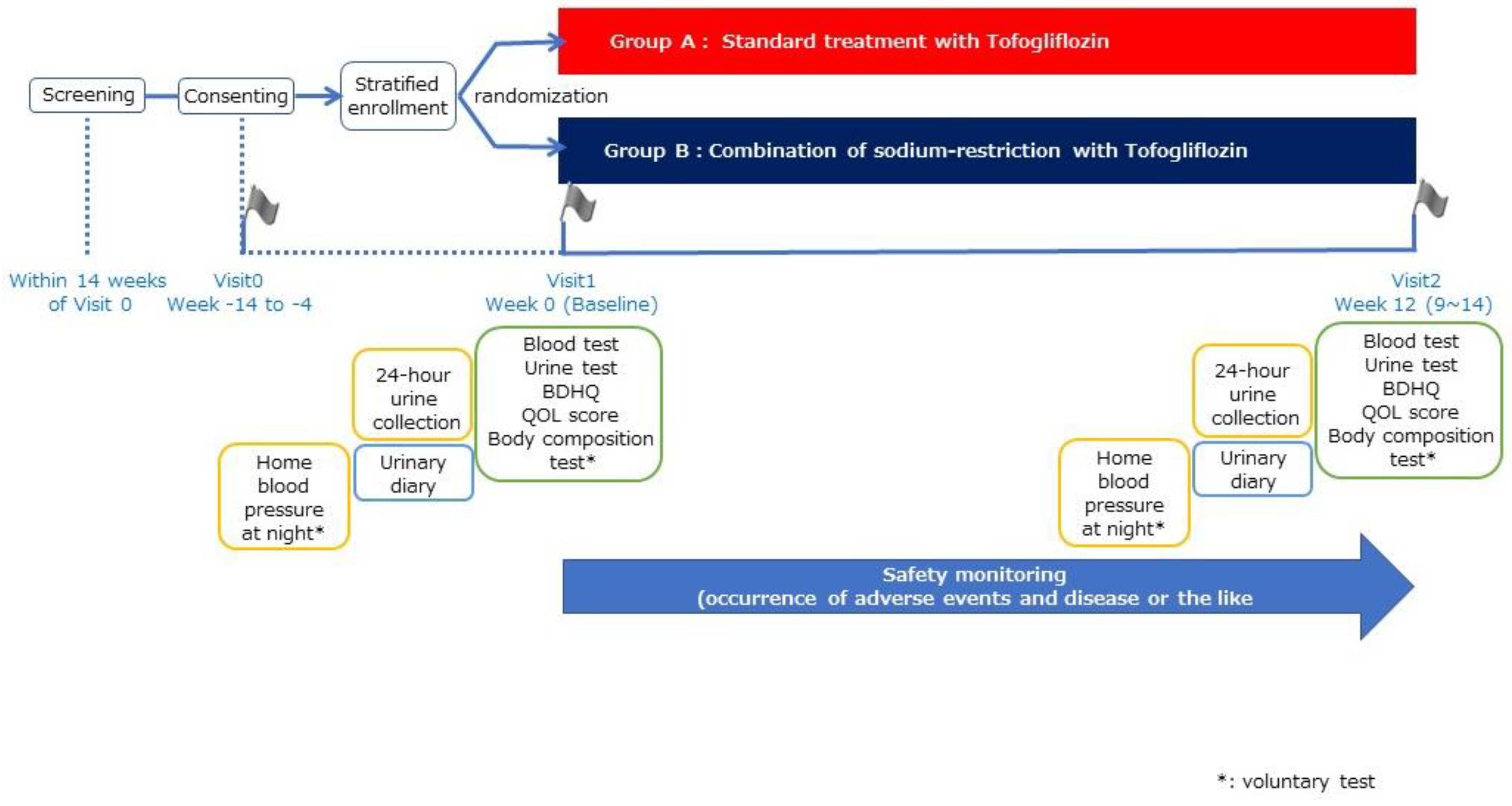
2.1. Trial Design
2.2. Eligibility Criteria, Recruitment, and Sample Size
2.3. Interventions
2.3.1. Random Grouping and Intervention Description
2.3.2. Details of Instruction on Sodium Restriction
2.3.3. Details of Other Dietary Instruction
2.3.4. Observation Items
2.4. Criteria for Discontinuing or Modifying Allocated Interventions
2.4.1. Criteria and Coping Strategies for Study Discontinuation
2.4.2. Criteria for Discontinuation of Study in Each Participant
2.4.3. Criteria for Discontinuation of Study
2.4.4. Coping with Adverse Events
2.5. Deviation from the Protocol
2.6. Management of Incompatibility
- When the responsible investigator becomes aware of the incompatibility of present study, the responsible investigator must immediately report this fact to the principal investigator.
- When the investigator becomes aware of the non-conformity of the study, the investigator immediately reports this fact to the responsible investigator.
- When there are serious incompatibilities, the investigator must immediately ask the accreditation review committee members for their opinions.
2.7. Strategies to Improve Adherence to Interventions
2.7.1. Management of the Study Agent
2.7.2. Outcomes
2.8. Data Collection and Management
2.8.1. Plans for Assessment and Collection of Outcomes
Original Documents
- Original documents for all data items (medical data, nurse records, drug records, laboratory data, subject logbooks, CRFs, QOL questionnaires, etc.).
- Records of informed consent indicating the patient’s agreement to the study participation.
2.8.2. Documents to Be Served as Source Documents
- Withdrawal of consent form.
- Medication adherence information.
- Adverse event and disease information.
2.8.3. Data Management and Confidentiality
2.9. Patient and Public Involvement Statement
2.10. Statistical Methods
2.10.1. Analysis of the Primary Endpoint
2.10.2. Analysis of the Secondary Endpoints
2.10.3. Methods for Additional Analyses (e.g., Subgroup Analyses)
2.11. Ethics and Dissemination
2.11.1. Data Handling Committee
2.11.2. Composition of the Data Monitoring Committee, Its Role, and Reporting Structure
2.12. Adverse Event Reporting and Harms
2.12.1. Reporting of Adverse Events
2.12.2. Frequency and Plans for Auditing Trial Conduct
2.12.3. Dissemination Plans
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abrams, P.; Cardozo, L.; Fall, M.; Griffiths, D.; Rosier, P.; Ulmsten, U.; van Kerrebroeck, P.; Victor, A.; Wein, A. The standardisation of terminology of lower urinary tract function: Report from the standardisation sub-committee of the international continence society. Am. J. Obstet. Gynecol. 2002, 187, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Yokoyama, O. Pathogenesis and management of nocturia in the elderly. Jpn. J. Geriatr. 2013, 50, 434–439. (In Japanese) [Google Scholar]
- Yoshimura, K.; Terada, N.; Matsui, Y.; Terai, A.; Kinukawa, N.; Arai, Y. Prevalence of and risk factors for nocturia: Analysis of a health screening program. Int. J. Urol. 2004, 11, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, H.; Xu, K.; Zhang, X.; Wang, X.; Na, Y.; Kang, X. Prevalence, risk factors, and symptom bother of nocturia: A population-based survey in China. World J. Urol. 2015, 33, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wen, Y.B.; Wang, Z.M.; Wen, J.G.; Li, Z.Z.; Shang, X.P.; Liu, Z.S.; Jia, L.H.; Qin, G.J.; Heesakkers, J.; et al. Risk Factors of Nocturia (Two or More Voids Per Night) in Chinese People Older Than 40 Years. Neurourol. Urodyn. 2015, 570, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Madhu, C.; Coyne, K.; Hashim, H.; Chapple, C.; Milsom, I.; Kopp, Z. Nocturia: Risk factors and associated comorbidities; findings from the EpiLUTS study. Int. J. Clin. Pr. 2015, 69, 1508–1516. [Google Scholar] [CrossRef]
- Chow, P.M.; Liu, S.P.; Chuang, Y.C.; Lee, K.S.; Yoo, T.K.; Liao, L.; Wang, J.; Liu, M.; Sumarsono, B.; Jong, J.J. The prevalence and risk factors of nocturia in China, South Korea, and Taiwan: Results from a cross-sectional, population-based study International Index of Erectile Function. World J. Urol. 2018, 36, 1853–1862. [Google Scholar] [CrossRef]
- Hirayama, A.; Torimoto, K.; Mastusita, C.; Okamoto, N.; Morikawa, M.; Tanaka, N.; Yoshida, K.; Fujimoto, K.; Hirao, Y.; Kurumatani, N. Evaluation of Factors Influencing the Natural History of Nocturia in Elderly Subjects: Results of the Fujiwara-kyo Study. J. Urol. 2013, 189, 980–986. [Google Scholar] [CrossRef]
- Furukawa, S. Smoking and prevalence of nocturia in Japanese patients with type 2 diabetes mellitus: A post-hoc analysis of The Dogo Study. Neurourol. Urodyn. 2017, 36, 1336–1341. [Google Scholar] [CrossRef]
- Kawano, R.; Takahashi, F.; Hashimoto, Y.; Okamura, T.; Miki, A.; Kaji, A.; Sakai, R.; Kitagawa, N.; Senmaru, T.; Majima, S.; et al. Short energy intake is associated with muscle mass loss in older patients with type 2 diabetes: A prospective study of the KAMOGAWA-DM cohort. Clin. Nutr. 2021, 40, 1613–1620. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Sakai, R.; Ikeda, K.; Fukui, M. Association between sleep disorder and quality of life in patients with type 2 diabetes: A cross-sectional study. BMC Endocr. Disord. 2020, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Niu, K.; Hozawa, A.; Ikeda, Y.; Kaiho, Y.; Ohmori-Matsuda, K.; Nakaya, N.; Kuriyama, S.; Ebihara, S.; Nagatomi, R.; et al. Impact of Nocturia on Bone Fracture and Mortality in Older Individuals: A Japanese Longitudinal Cohort Study. J. Urol. 2010, 184, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.C.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- The Japan Diabetes Society. Recommendations for the Appropriate Use of SGLT2 Inhibitors. Available online: http://www.fa.kyorin.co.jp/jds/uploads/recommendation_SGLT2.pdf (accessed on 22 February 2022). (In Japanese).
- Takegoshi, S.; Miyata, Y.; Ogata, N.; Saito, T. Availability and efficacy of 20-mg tofogliflozin administered every other day to type 2 diabetic patients. Prog. Med. 2015, 35, 1077–1088. (In Japanese) [Google Scholar]
- Takeishi, S.; Tsuboi, H.; Takekoshi, S. Comparison of tofogliflozin 20 mg and ipragliflozin 50 mg used together with insulin glargine 300 U/ml using continuous glucose monitoring (CGM): A randomized crossover study. Endocr. J. 2017, 64, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasoe, S.; Maruguchi, Y.; Kajiya, S.; Uenomachi, H.; Miyata, M.; Kawasoe, M.; Kubozono, T.; Ohishi, M. Mechanism of the blood pressure-lowering effect of sodium-glucose cotransporter 2 inhibitors in obese patients with type 2 diabetes. BMC Pharmacol. Toxicol. 2017, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Miyata, Y.; Sakai, H. Effect of salt intake reduction on nocturia in patients with excessive salt intake. Neurourol. Urodyn. 2019, 38, 927–933. [Google Scholar] [CrossRef]
- Ushigome, E.; Oyabu, C.; Iwai, K.; Kitagawa, N.; Kitae, A.; Kimura, T.; Yokota, I.; Ushigome, H.; Hamaguchi, M.; Asano, M.; et al. Effects of dietary salt restriction on home blood pressure in diabetic patients with excessive salt intake: A pilot study. J. Clin. Biochem. Nutr. 2019, 65, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Miyata, Y.; Sakai, H. Daily salt intake is an independent risk factor for pollakiuria and nocturia. Int. J. Urol. 2017, 24, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Tabara, Y.; Murase, K.; Setoh, K.; Kawaguchi, T.; Nagashima, S.; Kosugi, S.; Nakayama, T.; Wakamura, T.; Hirai, T.; et al. Nocturia and increase in nocturnal blood pressure: The Nagahama study. J. Hypertens. 2018, 36, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Obayashi, K.; Tai, Y.; Yamagami, Y.; Negoro, H.; Kataoka, H.; Kurumatani, N.; Saeki, K. Association between NT-proBNP and nocturia among community-dwelling elderly males and females: A cross-sectional analysis of the HEIJO-KYO study. Neurourol. Urodyn. 2021, 40, 112–119. [Google Scholar] [CrossRef]
- Ishii, H. The Japanese version of the Diabetes Treatment Satisfaction Questionnaire (DTSQ): Translation and clinical evaluation. J. Clin. Exp. Med. 2000, 192, 809–814. [Google Scholar]
- Sato, E.; Ochiai, R.; Shibayama, T.; Nishigaki, M.; Abe, Y.; Sawa, T.; Suzukamo, Y.; Kazuma, K. Reliability and validity of revised and short form versions of diabetes diet-related quality of life scale. Diabetol. Int. 2017, 8, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Honda, S.; Murakami, K.; Sasaki, S.; Okubo, H.; Hirota, N.; Notsu, A.; Fukui, M.; Date, C. Both comprehensive and brief self-administered diet history questionnaires satisfactorily rank nutrient intakes in Japanese adults. J. Epidemiol. 2012, 22, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Homma, Y.; Yoshida, M.; Yamanishi, T.; Gotoh, M. Core lowerurinary tract symptom (CLSS) questionnaire: A reliable tool in the overall assessment of lower urinary tract symptoms. Int. J. Urol. 2008, 15, 816–820. [Google Scholar] [CrossRef]


{kind=link}
| Inclusion criteria | |
| Patients with all of the following criteria will be considered | |
| 1 | Patients with type 2 diabetes |
| 2 | Patients with nocturia more than once |
| 3 | Male and female between the ages of 20 and 90 at the time of obtaining consent |
| 4 | Patients who provide their consent in a written form |
| Exclusion criteria | |
| Patients who meet any of the following criteria are not eligible to the study | |
| 1 | Patients who use SGLT2 inhibitor at least 3 months prior to giving their content |
| 2 | Patients who have already been instructed by a nutritionist of sodium-restriction |
| 3 | Patients with an estimated sodium intake of less than 6 g/day by urinalysis at the time of obtaining consent |
| 4 | Patients for whom tofogliflozin is contraindicated |
| 5 | Patients whose HbA1c is 10.5% or higher within 3 months |
| 6 | Patients with eGFR less than 15 mL/min/1.73 m2 or serum creatinine higher than 3.5 mg/dL or with hemodialysis |
| 7 | Patients with low blood pressure (less than 100/60 mmHg) |
| 8 | Patients with unstable hypertension |
| 9 | Patients with activities of daily living (ADL) of PS2 or higher |
| 10 | Patients with heart failure classified as New York Heart Association (NYHA) category Ⅲ or Ⅳ |
| 11 | Patients being pregnant or planning to become pregnant. |
| 12 | Patients suffering from cancer. However, the treatment has been completed and/or the cancer has not recurred and/or is becoming apparent |
| 13 | Patients with anemia (Hb is 10 g/dL or less) caused by primary diseases other than diabetic nephropathy |
| 14 | Patients with hypoalbuminemia (serum albumin is 3.5 g/dL or less) caused by primary diseases other than diabetic nephropathy |
| 15 | Patients with nephrotic syndrome (less than 3.0 g/dL of serum albumin and more than 3.5 g/day of urinary protein) due to primary disease except diabetic nephropathy |
| 16 | Patients judged to be non-adherent by the attending physician |
| 17 | Patients who require substitute to obtain consent |
| 18 | Patients deemed inappropriate by the attending physician |
| 1. Eligibility information | |
| Observation point | At consenting and enrollment |
| Observation item | Inclusion criteria, exclusion criteria, gender, age, date of giving consent |
| 2. Background information | |
| Observation point | At consenting, enrollment, or week 0 |
| Observation item | Gender, age, height, weight, body mass index, duration of diabetes, presence of comorbidities (presence/absence of macrovascular/microvascular complications, renal disease, hepatic disease, hypertension, dyslipidemia), and history of illness (history of cardio-cerebrovascular disease) |
| 3. Medication information | |
| Observation point | Week 0 and week 12 |
| Observation item | Medications for diabetes, daily dosage, and other concomitant medication |
| 4. Blood tests (fasting) | |
| Observation point | Week 0 and week 12 |
| Observation item | Red blood cell count, white blood cell count, hematocrit, hemoglobin, estimated plasma volume, blood platelet count, hepatic enzymes (AST, ALT, serum albumin, LDH, gamma-GTP, ALP), UA, BUN, Cre, eGFR, T-Chol, HDL, LDL, TG, serum Na/K/Cl, HbA1c (or glycoalbumin), and plasma glucose |
| 5. Special blood tests (fasting, using residual sample of “6 blood tests”) | |
| Observation point | Week 0 and week 12 |
| Observation item | Total ketone body, beta-hydroxybutyric acid, acetoacetic acid, and plasma metabolome++ |
| 6. Urine tests (total) | |
| Observation point | Week 0 and week 12 |
| Observation item | Specific gravity, pH, protein, glucose, ketone body, occult blood, bilirubin, urobilinogen, u-mAlb, U-Cre, and U-Na/K/Cl |
| 7. Body composition test (optional test) | |
| Observation point | Week 0 and week 12 |
| Observation item | Skeletal muscle mass, skeletal muscle mass to total body weight ratio, and fat mass |
| 8. QOL score (questionnaire to whom the participants directly answer) | |
| Observation point | Week 0 and week 12 |
| Observation item |
|
| 9. Other items that the participants should measure on their own: Urinary diary | |
| Observation point | Week 0: conducted within 7 days after obtaining consent until the observation period week 0. Urine volume will be measured for 3 days; week 12: conducted within 7 days before observation point week 12. Urine volume will be measured for 3 days. |
| Observation item | Time of going to bed, time of waking up, time of urination, frequency of urination, volume of urine, and alcohol consumption The study participants store urine using a measuring cup. |
| 10. 24 h urine collection test | |
| Observation point | Week 0 (24 h before the observation point) and week 12 (24 h before the observation point) |
| Observation item | Time of going to bed, time of waking up, volume of urine, volume of urine at night, plasma glucose, urinary creatinine excretion for 24 h, and urinary sodium excretion for 24 h The study participants store urine using a measuring cup and dispense a small portion of this urine into a spit. |
| 11. Home blood pressure at night | |
| Observation point | Week 0 (5 days before observation point. Urine volume will be measured 3 times a day for 3 days), week 12 (conducted within 7 days before observation point week 12, 3 times each day) Conducted in a period that is different from the urinary diary period |
| Observation item | Home blood pressure at night: The participants measure their blood pressure three times at night using an upper arm blood pressure monitor (Omron HEM-9601T) (automatic measurement). The participants record the results in their diaries. |
| 12. Adherence to research medication regimen and sodium restriction | |
| Observation point | Week 12 |
| Observation item | The research physician conducts an interview to collect data regarding the patient’s medication status and adherence with sodium restriction, and record the collected data in the CRF. |
| Observation Items | Enrollment | Baseline Week 0 *1 | Week 12 (Week 9–14) or at Discontinuation *2 |
|---|---|---|---|
| Obtaining consent | ◯ | ||
| ① Eligibility information | ◯ | ||
| ② Background information | ◯ | ||
| ③ Medication information | ◯ | ◯ | |
| ④ Blood tests | ◯ | ◯ | |
| ⑤ Special blood tests | ◯ | ◯ | |
| ⑥ Urine tests | ◯ | ◯ | |
| ⑦ Body composition | ▲ | ▲ | |
| ⑧ Questionnaire | ◯ | ◯ | |
| ⑨ Urination log | ◯ | ◯ | |
| ⑩ 24 h urine collection | ◯ | ◯ | |
| ⑪ Nocturnal home blood pressure | ▲ | ▲ | |
| ⑫ Compliance with drug regimen/Compliance with sodium restriction | ◯ | ||
| ⑬ Adverse event and disease or the like | ←○→ | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakajima, H.; Okada, H.; Kogure, A.; Osaka, T.; Tsutsumi, T.; Tanaka, T.; Hasegawa, G.; Mogami, S.; Mitsuhashi, K.; Kitagawa, N.; et al. Multicenter, Open Label, Randomized Controlled Superiority Trial for Availability to Reduce Nocturnal Urination Frequency: Study Protocol for a TOP-STAR Study. Diabetology 2022, 3, 620-633. https://doi.org/10.3390/diabetology3040048
Nakajima H, Okada H, Kogure A, Osaka T, Tsutsumi T, Tanaka T, Hasegawa G, Mogami S, Mitsuhashi K, Kitagawa N, et al. Multicenter, Open Label, Randomized Controlled Superiority Trial for Availability to Reduce Nocturnal Urination Frequency: Study Protocol for a TOP-STAR Study. Diabetology. 2022; 3(4):620-633. https://doi.org/10.3390/diabetology3040048
Chicago/Turabian StyleNakajima, Hanako, Hiroshi Okada, Akinori Kogure, Takafumi Osaka, Takeshi Tsutsumi, Toru Tanaka, Goji Hasegawa, Shinichi Mogami, Kazuteru Mitsuhashi, Noriyuki Kitagawa, and et al. 2022. "Multicenter, Open Label, Randomized Controlled Superiority Trial for Availability to Reduce Nocturnal Urination Frequency: Study Protocol for a TOP-STAR Study" Diabetology 3, no. 4: 620-633. https://doi.org/10.3390/diabetology3040048