
Glucocorticoid Receptor Blockers Pretreatment Did Not Improve Infarct Volume in Type-2 Diabetic Mouse Model of Stroke
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model of Hypoxia/Ischemia
2.2. Glucocorticoid Receptor Blocker Treatment
2.3. Histological Analysis of Stroke
2.4. Corticosterone Measurement
2.5. Statistical Analysis
3. Results
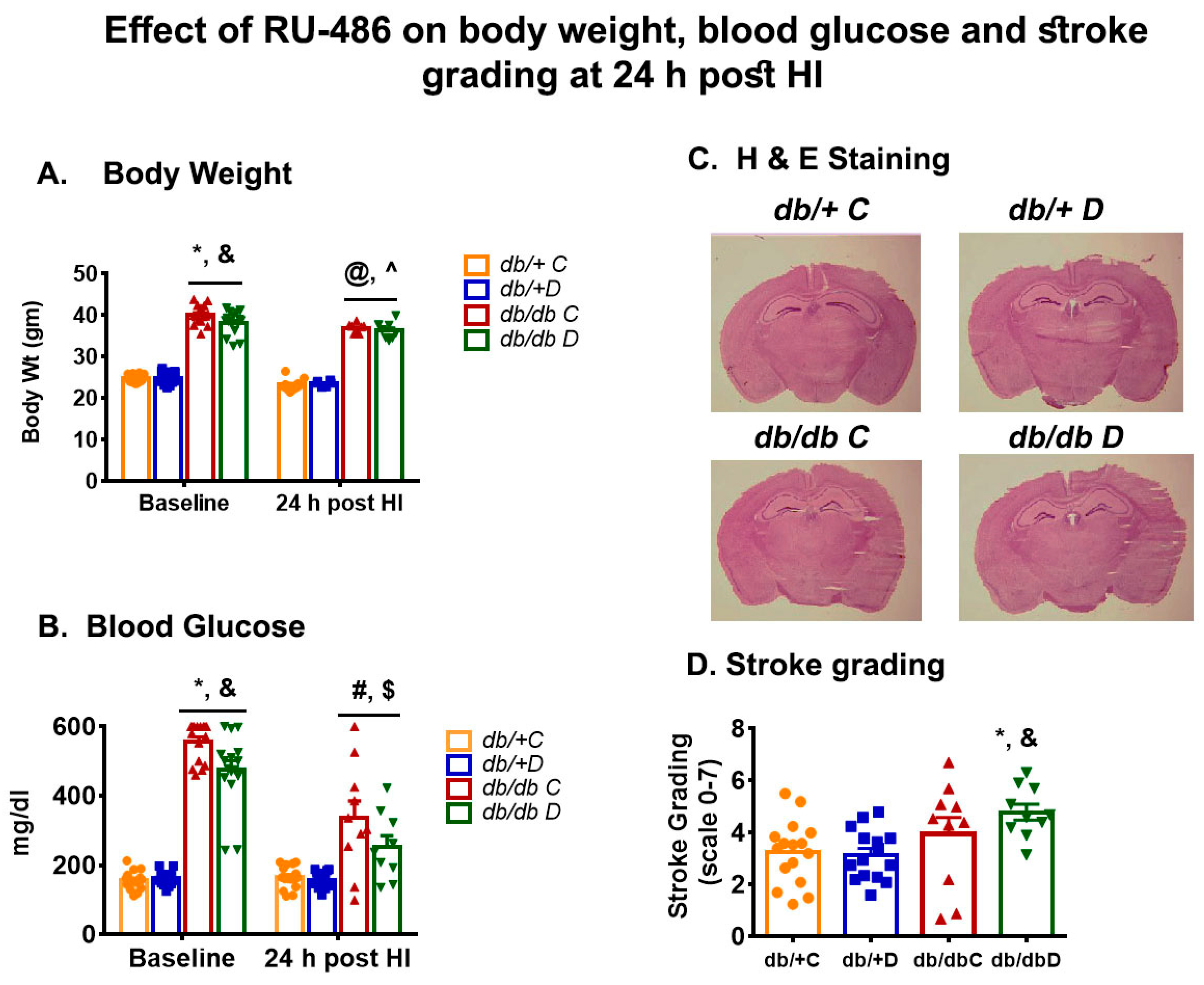
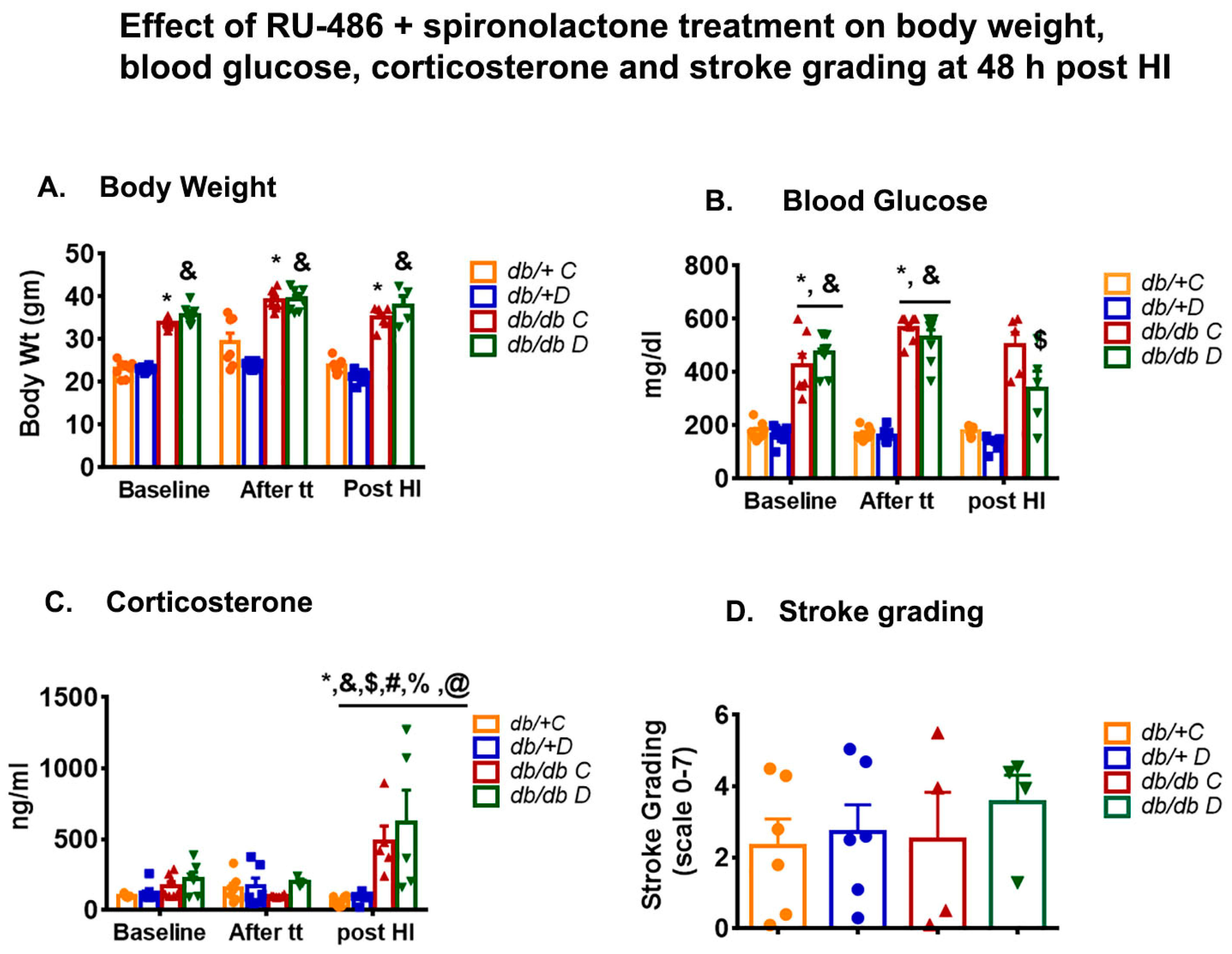
3.1. Effect of Glucocorticoid Receptor Blockers on Body Weight and Blood Glucose
3.2. Effect of Glucocorticoid Receptor Blockers on Corticosterone
3.3. Effect of Glucocorticoid Receptor Blockers on Infarct Size
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.; Park, E.S.; Chen, P.R.; Kim, E. Dysregulated Hypothalamic-Pituitary-Adrenal Axis Is Associated With Increased Inflammation and Worse Outcomes After Ischemic Stroke in Diabetic Mice. Front Immunol. 2022, 13, 864858. [Google Scholar] [CrossRef] [PubMed]
- Stuller, K.A.; Jarrett, B.; DeVries, A.C. Stress and social isolation increase vulnerability to stroke. Exp. Neurol. 2012, 233, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, E.; Beltran, C.; Cho, S. Corticosterone-Mediated Body Weight Loss Is an Important Catabolic Process for Poststroke Immunity and Survival. Stroke 2019, 50, 2539–2546. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.; Boysen, G.; Johannesen, H.H. Serum-cortisol reflects severity and mortality in acute stroke. J. Neurol. Sci. 2004, 217, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Marklund, N.; Peltonen, M.; Nilsson, T.K.; Olsson, T. Low and high circulating cortisol levels predict mortality and cognitive dysfunction early after stroke. J. Intern. Med. 2004, 256, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mracsko, E.; Liesz, A.; Karcher, S.; Zorn, M.; Bari, F.; Veltkamp, R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav. Immun. 2014, 41, 200–209. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Cortisol-induced insulin resistance in man: Impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J. Clin. Endocrinol. Metab. 1982, 54, 131–138. [Google Scholar] [CrossRef]
- Friedman, J.E.; Yun, J.S.; Patel, Y.M.; McGrane, M.M.; Hanson, R.W. Glucocorticoids regulate the induction of phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J. Biol. Chem. 1993, 268, 12952–12957. [Google Scholar] [CrossRef]
- Shimomura, Y.; Bray, G.A.; Lee, M. Adrenalectomy and steroid treatment in obese (ob/ob) and diabetic (db/db) mice. Horm. Metab. Res. 1987, 19, 295–299. [Google Scholar] [CrossRef]
- Saito, M.; Bray, G.A. Adrenalectomy and food restriction in the genetically obese (ob/ob) mouse. Am. J. Physiol. Integr. Comp. Physiol. 1984, 246, R20–R25. [Google Scholar] [CrossRef] [PubMed]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Tseng, M.T. Preischemic hyperglycemia-aggravated damage: Evidence that lactate utilization is beneficial and glucose-induced corticosterone release is detrimental. J. Neurosci. Res. 2001, 66, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.S.; Tseng, M.T.; Schurr, A. The glucose paradox of cerebral ischemia: Evidence for corticosterone involvement. Brain Res. 2003, 971, 9–17. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Stress, Glucocorticoids, and Damage to the Nervous System: The Current State of Confusion. Stress 1996, 1, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Leng, T.; Yang, T.; Zeng, Z.; Ueki, T.; Xiong, Z.G. Role of serum- and glucocorticoid-inducible kinases in stroke. J. Neurochem. 2016, 138, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Bertorelli, R.; Adami, M.; Di Santo, E.; Ghezzi, P. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci. Lett. 1998, 246, 41–44. [Google Scholar] [CrossRef]
- Smith-Swintosky, V.L.; Pettigrew, L.C.; Sapolsky, R.M.; Phares, C.; Craddock, S.D.; Brooke, S.M.; Mattson, M.P. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J. Cereb. Blood Flow Metab. 1996, 16, 585–598. [Google Scholar] [CrossRef]
- Bettermann, K.; Sinha, K.; Kumari, R.; Fox, C.; Simpson, I.A. The peripheral immune response in hyperglycemic stroke. Clin. Neurol. Neurosurg. 2020, 195, 106061. [Google Scholar] [CrossRef]
- Kumari, R.; Willing, L.B.; Krady, J.K.; Vannucci, S.J.; Simpson, I.A. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J. Cereb. Blood Flow Metab. 2007, 27, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Willing, L.B.; Patel, S.D.; Krady, J.K.; Zavadoski, W.J.; Gibbs, E.M.; Vannucci, S.J.; Simpson, I.A. The PPAR-gamma agonist, darglitazone, restores acute inflammatory responses to cerebral hypoxia-ischemia in the diabetic ob/ob mouse. J. Cereb. Blood Flow Metab. 2010, 30, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Figlewicz, D.P.; Baskin, D.G.; Woods, S.C.; Porte, D., Jr. Insulin in the brain: A hormonal regulator of energy balance. Endocr. Rev. 1992, 13, 387–414. [Google Scholar] [PubMed]
- Freedman, M.R.; Horwitz, B.A.; Stern, J.S. Effect of adrenalectomy and glucocorticoid replacement on development of obesity. Am. J. Physiol. Content 1986, 250, R595–R607. [Google Scholar] [CrossRef]
- Björntorp, P.; Rosmond, R. Obesity and cortisol. Nutrition 2000, 16, 924–936. [Google Scholar] [CrossRef]
- Liu, Y.; Nakagawa, Y.; Wang, Y.; Sakurai, R.; Tripathi, P.V.; Lutfy, K.; Friedman, T.C. Increased glucocorticoid receptor and 11{beta}-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes 2005, 54, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Willing, L.B.; Patel, S.D.; Baskerville, K.A.; Simpson, I.A. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J. Neurochem. 2011, 119, 1029–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Willing, L.; Kimball, S.R.; Simpson, I.A. The effect of chronic exposure to metformin in a new type-2 diabetic NONcNZO10/LtJ mouse model of stroke. Pharmacol. Rep. 2022, 74, 696–708. [Google Scholar] [CrossRef]
- Kumari, R.; Willing, L.B.; Jefferson, L.S.; Simpson, I.A.; Kimball, S.R. REDD1 (regulated in development and DNA damage response 1) expression in skeletal muscle as a surrogate biomarker of the efficiency of glucocorticoid receptor blockade. Biochem. Biophys. Res. Commun. 2011, 412, 644–647. [Google Scholar] [CrossRef] [Green Version]
- Martín, A.; Rojas, S.; Chamorro, A.; Falcón, C.; Bargalló, N.; Planas, A.M. Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein O-glycosylation. Stroke 2006, 37, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Glezer, I.; Rivest, S. Glucocorticoids: Protectors of the brain during innate immune responses. Neuroscientist 2004, 10, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Soulet, D.; Rivest, S. Polyamines play a critical role in the control of the innate immune response in the mouse central nervous system. J. Cell Biol. 2003, 162, 257–268. [Google Scholar] [CrossRef]
- Nadeau, S.; Rivest, S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J. Neurosci. 2003, 23, 5536–5544. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Treatment Stages | Study 1 (RU-486, 40 mg/kg; I.P.) | Study 2 (RU-486+ Spironolactone, 25 mg/kg; S.C.) |
|---|---|---|
| First dose | 24 h prior to surgery | daily injection 1 week prior to surgery |
| Second dose | just after surgery, | just after surgery |
| Third dose | 7 h post-HI, | 24 post-HI |
| Sample Collection | 24 h post-HI | 48 h post-HI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, R.; Willing, L. Glucocorticoid Receptor Blockers Pretreatment Did Not Improve Infarct Volume in Type-2 Diabetic Mouse Model of Stroke. Diabetology 2022, 3, 539-548. https://doi.org/10.3390/diabetology3040041
Kumari R, Willing L. Glucocorticoid Receptor Blockers Pretreatment Did Not Improve Infarct Volume in Type-2 Diabetic Mouse Model of Stroke. Diabetology. 2022; 3(4):539-548. https://doi.org/10.3390/diabetology3040041
Chicago/Turabian StyleKumari, Rashmi, and Lisa Willing. 2022. "Glucocorticoid Receptor Blockers Pretreatment Did Not Improve Infarct Volume in Type-2 Diabetic Mouse Model of Stroke" Diabetology 3, no. 4: 539-548. https://doi.org/10.3390/diabetology3040041