
Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes


1
Department of Immunology, University of Pittsburgh, Pittsburgh, PA 15261, USA
2
Novasenta Inc., Pittsburgh, PA 15219, USA
Diabetology 2022, 3(1), 79-96; https://doi.org/10.3390/diabetology3010007
Submission received: 27 December 2021
/
Revised: 8 January 2022
/
Accepted: 20 January 2022
/
Published: 7 February 2022
(This article belongs to the Special Issue Diabetology: Feature Papers 2021)
Abstract
:Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of insulin-producing pancreatic β-cells by their own immune system, resulting in lifelong insulin deficiency. Continuous exogenous insulin replacement therapy is the current standard of care for T1D. Transplantation of primary pancreatic islets or the entire pancreas is a viable remedy for managing patients with autoimmune T1D. However, this strategy is not feasible due to several obstacles, including a scarcity of donors, islet cells, and poor vascular engraftment of islets post-transplantation, as well as the need for prolonged immune suppression. Innovative approaches must be developed to counteract pancreatic β-cell destruction and salvage endogenic insulin production, thereby regulating blood glucose levels. This review includes an overview of autoimmune T1D, immune cells involved in T1D pathophysiology, and immunotherapy-based strategies to treat and prevent autoimmune T1D. Recent immunotherapy progress toward targeting pancreatic islet-specific immune pathways tangled tolerance has fueled the advancement of therapies that may allow for the prevention or reversal of this autoimmune T1D while avoiding other adverse reactions associated with the previous attempt, which was mostly immunosuppressive. As a result, significant efforts are currently underway to improve the efficacy of immunotherapy-based approaches by leveraging the beneficial actions of immune cells, specifically effector CD4+, CD8+, and regulatory T cells. This review will provide an overview of currently available immune-based therapeutic options for T1D and will examine the growing evidence that supports the use of immune cell-based approaches to improve therapeutic outcomes in the prevention or reversal of autoimmune T1D.
1. Introduction
Autoimmune diseases, which are pathological conditions caused by abnormal immune responses to substances and tissues that are normally present or generated within their own body, affect nearly 23.5 million Americans, with nearly 80 percent of those affected being females. Around 100 or more autoimmune diseases have been reported, according to the American Autoimmune Related Diseases Association (AARDA). Many of them have similar symptoms, making them more difficult to diagnose and distinguish. Autoimmune diseases can affect almost any organ in the body, including the heart, muscles, eyes, brain, nerves, skin, joints, lungs, kidneys, glands, the digestive tract, and blood vessels. Although there are over 100 autoimmune diseases known to date, the most common autoimmune disorders are Type 1 Diabetes Mellitus (T1D), Rheumatoid Arthritis (RA), Systemic Lupus Erythematosus (SLE), Inflammatory Bowel Disease (IBD), Multiple Sclerosis (MS), Guillain-Barre syndrome, chronic inflammatory demyelinating polyneuropathy, Psoriasis, Graves’ disease, and Hashimoto’s disease [1,2].
As we all know, one of the immune system’s most important functions is to protect the body by responding to attacking pathogens such as bacteria, parasites, or viruses by producing antibodies or sensitized lymphocytes to fight them. An immune response against one’s own body cannot be triggered or tolerated in healthy conditions. However, in some cases, the body’s immune cells or tissue make a mistake and behave as if they are foreign because our immune system recognizes them as foreign entities and attacks their own body rather than protecting them. These irrational responses can result in a variety of autoimmune diseases. T1D, also known as Juvenile Diabetes, is a chronic autoimmune disease in which both T and B lymphocytes initiate a specific targeted immune response that leads to the destruction of insulin-producing pancreatic β-cells [3,4,5].
According to the Centers for Disease Control and Prevention, approximately 10% of the American population has diabetes, with type 1 diabetes accounting for 5–10% of those affected (T1D). The number of children diagnosed with various autoimmune diseases, in addition to autoimmune T1D, has increased. T1D is one of the most common childhood chronic diseases, affecting approximately 70,000 children each year. Worldwide, 111,1100 young people under the age of 20 have autoimmune T1D, and its prevalence in children is increasing by 3–5 percent per year, which is far too high. Pediatric T1D prevalence rates vary by region, but are typically lower in Asian countries and higher in the rest of the world [6,7]. Many autoimmune disorders, including T1D, are caused by flaws in the regulation of effector immune cell populations, which may be due to malfunctions in the immune-mediated suppression or immunological tolerance mechanisms.
T1D is a complex autoimmune disease characterized by genomic, epigenomic, and environmental factors that influence both adaptive and innate effector immune cell populations, culminating in pathological, chronic islet inflammation. The heterogeneity associated with human autoimmune T1D, as well as the nature of islet inflammation, reflects the individual’s genotype and type of environmental insult. These factors influence which immune effectors are important in the pathophysiology of autoimmune T1D, the rate of disease progression, and the degree of pancreatic islet-specific β-cell dysregulation and/or death [8,9,10,11].
The emergence of autoimmune disorders resulted from the immune system’s inability to control autoreactive cascading responses. The ability to reprogram the immune system to maintain homeostasis without ongoing treatment is the holy grail of autoimmune therapies such as T1D. The current treatment regimen for T1D patients is largely based on exogenous insulin injections or insulin pumps, which are lifesaving but not curative. While insulin replacement is beneficial for improving glycol-metabolic control, it is limited by its inherent inability to replicate endogenous insulin’s biological functions; this puts T1D patients at risk for hypoglycemic episodes [12]. Due to the accelerated T1D disease, autoimmune T1D patients have a significantly reduced life expectancy. There are several ways to rebuild complete endocrine pancreatic function, but cadaveric whole pancreas or islets transplantation is only available to a small number of T1D patients. There was limited clinical success due to poor post-transplant engraftment of vascular cells, blood-mediated inflammatory response, hypoxia, hypoxia-reoxygenation injury, the alloimmune response against the graft, and persistent autoimmune attacks in T1D patients who received islets transplanted [13,14].
Several attempts have since been made to use emerging immunosuppressive approaches to provide immune protection to grafted islets or the entire pancreas. Traditional treatments for autoimmune T1D rely primarily on broad-spectrum immunosuppressive agents. They require much safer and more effective treatment due to the severe side effects of harsh immunosuppressive regimens.
Targeted and specific immunotherapies must be developed for the treatment of autoimmune T1D to re-establish tolerance mechanisms or eliminate pancreatic cell-specific immune responses generated by primary T and B lymphocytes, which can preserve the destructive β-cell mass for different functions and maintain insulin throughout life. Developing selective immunotherapies for autoimmune T1D will necessitate a thorough understanding of immunological responses to pancreatic cell-derived peptides, which serve as epitopes/antigens responsible for autoimmunity T1D, as well as how cognate T and B cells avoid tolerance checkpoints. This review discusses the general history of T1D, immune cells involved in the pathophysiology of autoimmune Type 1 diabetes, and immunotherapy-based treatment strategies for T1D.
2. Background of Autoimmune T1D
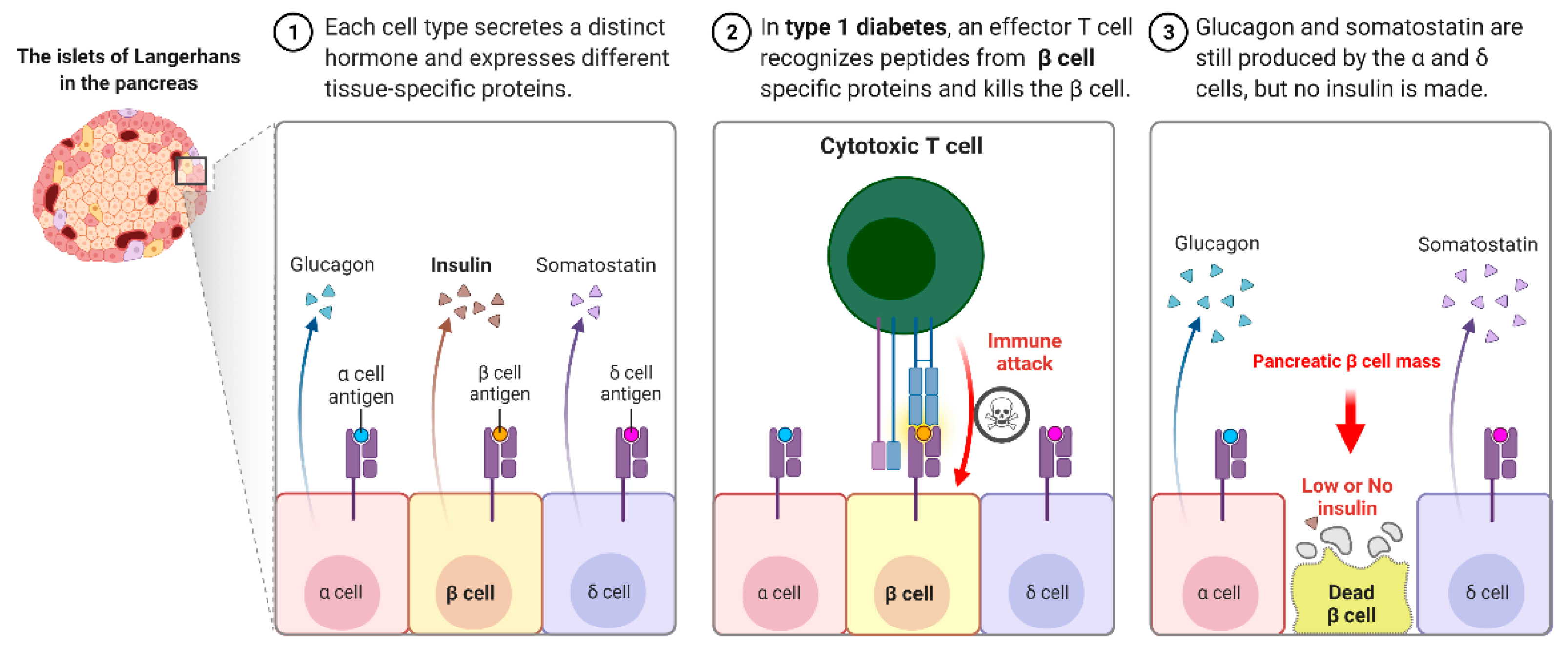
Autoimmune T1D occurs when our own immune system mistakenly destroys the insulin hormone-producing pancreatic β-cells in the Langerhans islets, schematically shown in Figure 1. T1D appears to have a genetic component and can be diagnosed in childhood and reported in later life, i.e., adulthood. Its causes are not completely understood or researched, and there is currently no likely treatment or cure. To survive, T1D patients must rely on injected or pumped insulin for the rest of their lives. T1D is found in both children and adults who exhibit symptoms such as frequent urination, dry mouth, increased thirst, itchy or dry skin, increased appetite, unexplained weight loss, and infections. Individuals with autoimmune T1D are typically diagnosed after exhibiting symptoms such as nausea, vomiting, extreme thirst, exhaustion, and/or malaise. As the body loses its ability to produce insulin, a hormone that allows the body to use the sugar found in everyday foods, known as glucose, as an energy source, patients with T1D must work closely with their endocrinologists to determine the insulin doses and lifestyle changes needed to manage their blood sugar levels. Autoimmune T1D patients are vulnerable to a variety of health issues, ranging from minor to severe; if you are not properly managed, your glucose levels will rise. Most T1D patients spend their lives managing it and have a problem with it, blood glucose levels are outside the clinically recommended beneficial range, which leads to potentially fatal hyperglycemia (high blood glucose) episodes and hypoglycemia (low blood glucose). Assume you do not maintain it and develop high blood sugar, which frequently leads to devastating health complications later in life, such as blindness, kidney failure, heart disease, and nerve damage, resulting in amputations.
Several factors are responsible for the causes of T1D; the scientific community does not distinguish between the exact causes of autoimmune T1D, but they do see some onset factors and triggers associated with the condition. There is frequently a family history of T1D, and the best diagnoses occur in people who have no family members with the disease. Having a family history of autoimmune T1D, on the other hand, puts people at an increased risk of developing the disease. Some scientists believe viral infections can cause or contribute to the onset of the disease. To clarify, developing a viral-based vaccine may be one method of preventing T1D. Some environmental factors may be linked to T1D, most likely because of changes in our environment, possibly via a few viral infections or other similar agents. Early exposure to those factors after birth may be linked to the development of autoimmune T1D. T1D is not caused by diet or lifestyle, but several studies point to genetics [15,16,17,18], ethnicity, age, and the likelihood of developing T1D; the presence of specific genes appears in many autoimmune T1D patients. As T1D does not discriminate, if someone in your family has had autoimmune T1D, you are more likely to develop the disease. Assume that people of all ethnic backgrounds have T1D, even though the prevalence increases in populations north of the equator. However, an individual can develop autoimmune T1D at any age, with the majority of cases being diagnosed in early elementary school or as preteens. Hormonal changes, like those associated with growth spurts, can have an impact on the presentation and management of T1D.
2.1. Immune Cells Involved in the Pathophysiology of Autoimmune Type 1 Diabetes
Tolerance has two mechanisms: central and peripheral tolerance. Both contribute to the defense against autoimmunity (Figure 2). T1D autoimmune disease can be caused by critical insufficiencies or restrictions in peripheral (lymph nodes/circulation) and/or central tolerance mechanisms (bone marrow/thymus), presenting a therapeutic opportunity. In healthy people, our immune systems are perfectly balanced to distinguish which antigens are foreign and which are self-antigens. To control this process, our immune system has a tolerance mechanism that includes thymic (central) tolerance, which eliminates high-affinity auto-antigen-specific T lymphocytes and those that fail to recognize auto-antigens completely and excuses T lymphocytes that recognize auto-antigens with intermediate affinity. As the naive immune repertoire is positively selected on auto antigens, self-recognition is hard-wired in the immune system, blurring the distinction between unwanted autoimmunity and desired immunity. As recirculating lymphocytes are exposed to tissue antigens under non-inflammatory conditions, which result in a tolerant, anergic state, our immune system’s peripheral tolerance system usually keeps potentially auto-specific lymphocytes in check. Nonetheless, in the presence of danger signals, such as infection and tissue damage, self-tolerance can be broken down, and autoimmune disease can progress. The immune pathogenesis of autoimmune T1D begins with a breakdown in self-tolerance, which results in the destruction of pancreatic β-cells by T lymphocytes (Figure 2).
Similarly, unwanted autoimmunity and desired host anti-pathogen specific immunity are inextricably linked, as effector immune responses that affect inflammatory tissue damage are analogous to those that mediate effective host defense. As a result, immunotherapeutic regimens that target the immune system’s common pathways invariably have both desired and unintended consequences. Several strategies for eliminating auto-antigen-specific immune cells by breaking tolerance to self-antigens can result in autoimmunity, which has been thoroughly reviewed [19]. Recent findings emphasize the importance of inhibitory receptors (IRs), which trigger or regulate autoimmunity’s peripheral tolerance. Later, deletion and blockade studies in animals and humans show a link to positive disease outcomes, highlighting the potential clinical benefits of enhancing IR signaling, specifically CTLA4, PD1, LAG3, TIM3, and TIGIT, to treat autoimmune diseases.
2.2. Immunotherapy-Based Approaches to Treating Autoimmune T1D
Drugs that target immune system elements provide relief for millions of people suffering from autoimmune diseases such as psoriasis and rheumatoid arthritis. Still, no single immunotherapy-based treatment for autoimmune T1D is currently approved. Few research organizations and pharmaceutical companies focus on multiple aspects of the immune system to develop an effective immune cell-based therapy to cure autoimmune T1D. Individuals with autoimmune T1D require lifetime medications in the form of regular insulin prescriptions to manage the condition, putting them at high risk of long-term complications. One day, the immunotherapy-based strategy will benefit T1D and become an insulin-free substitute to stop, prevent, and possibly cure this autoimmune T1D. They have focused on immune cell-based approaches for autoimmune T1D treatment since the 1980s, considering several possibilities such as the repair of unblanched immune tolerance, inhibition of diabetogenic T-cell or B-cell functionality, ex vivo regulatory T-cell (Treg) generation, repression of the innate arm of the immune system, inflammation, and HLA-matched islet transplantation rejection.
2.3. There Are a Few Obstacles to Immunotherapy-Based Treatment for Autoimmune T1D Cure
First, thanks to the boon of recombinant technology, insulin has long been available, so there has not been a pressing need for the development of new T1D therapies. T1D patients must check their blood sugar levels regularly and calculate the amount of insulin to inject. Still, let us assume they can keep this up indefinitely. In that case, it is possible to live a fairly normal life. Second, the primary reason for the lack of approved immunotherapies for T1D is that clinicians have been hesitant to refer patients with T1D to immunotherapy-based clinical trials. Due to endocrinologists’ lack of understanding, new-generation immunotherapies have fewer unpleasant and potentially dangerous side effects than older immune-suppressing drugs or immunotherapy only for oncology, as it was initially explored or assumed. Third, there has been a lack of engagement from pharmaceutical companies and research organizations, and fourth, compared to a disease like RA, people living with T1D represent a relatively small business discussed [20,21,22].
The following section of this review provides an overview and understanding of previous immunotherapy-based trials, describes recent and ongoing combination immunotherapy trials, and speculates on future combination clinical interventions to preserve insulin-secreting pancreatic-cells in autoimmune T1D.
The first immunotherapy trials for T1D were conducted more than 35 years ago. No single study has demonstrated clinically significant benefits from therapy with an acceptable risk profile [23]. A French Cyclosporine Diabetes Study was the first clinical trial to test the immunological intervention in type 1 diabetes [24]. Cyclosporine A (CSA) disrupts TCR-mediated signal transduction, inhibiting T-cell activation and T helper IL-2 secretion [25]. Later, a few studies demonstrated a significant reduction in exogenous insulin requirement after one year of immune-suppressive drug CSA treatment [24,26]. After CSA removal, blood glucose control worsens, and autoantibody levels are return [27].
Furthermore, cyclosporin medication has been linked to renal and pancreatic β-cell toxicity [27]. Despite the lack of long-term effects and potential toxicity, these trials heralded a new clinical era centered on immunomodulatory strategies to delay or prevent T1D. Numerous interventions, including parenteral insulin administration, dietary exposures, broad-spectrum immunosuppressants, anti-inflammatory drugs, and T- or B-cell targeted immunosuppressants, have been tested to date. At the same time, a small number of clinical trials have revealed modest benefits. There are no clinically approved immunotherapies for autoimmune T1D now, but a few developments are listed in Table 1 or highlighted in Figure 2. Let us look at a few of these immunotherapies and how they might help T1D patients.
Antibodies-based strategies: Suppression of effector or pancreatic β-cell attacking T-cells may aid in the protection of pancreatic β-cell destruction so that anti-T cell antibodies can be used. Based on the previous findings in the preclinical diabetic NOD mouse model, several clinical trials for T1D were conducted using a humanized monoclonal antibody against CD3 (anti-CD3, hOKT3 gamma1 (Ala-Ala), Teplizumab). Initially, two clinical trials lacked a placebo control group. They reported that endogenous C-peptide secretion was preserved while exogenous insulin requirements were reduced, with 5% of individuals achieving insulin independence for 48 weeks, which has been shown to preserve pancreatic β-cell function in people with T1D [46,47]. The most recent study demonstrates a delayed onset of T1D in high-risk individuals. According to a randomized, double-blind Phase II clinical trial found that a single 14-day regimen of Teplizumab (anti-CD3) delayed the onset of autoimmune T1D by 24.4 months when compared to a placebo-treated group and that 29 percent of treated patients had HbA1c levels less than 7% and insulin dose requirements less than 0.5 U/kg per day [48]. This study highlights its potential as a preventative treatment for autoimmune T1D. The FDA has granted anti-CD3 Ab teplizumab an advanced treatment designation to expedite the process of determining whether there are additional results to support its approval [49]. However, the cytokine storm caused significant side effects in the first version of the anti-CD3 drug.
Chemokines and cytokines regulate immune system function and cascading. Generally, when cytokines interact with their receptors expressed on various immune cells, complex signaling events occur, leading to the activation of downstream markers such as transcriptional factors. Later, these factors promote the activation and differentiation of immune cells into specific lineages, for example, naive CD4 T lymphocytes can differentiate into various subsets of CD4+ T cells (Th1, Th2, Th17, Treg, TFH, Tf9, etc.). This subset differentiation is influenced by the cytokine environment, as we know that an imbalance between Treg and Th17 leads to autoimmunity [50].
The combination of cytokines such as TGFβ and IL-6 with growth factor IL-2 regulates naïve T lymphocyte function and its fate toward developing either the Treg or Th17 lymphocytes, ultimately affecting the overall autoimmunity [51]. Both cytokine TGF-β and IL-6 facilitated the Th17 pathway by activating transcription factors STAT3 and RORγt, which are associated with the destruction of pancreatic β-cells in T1D [52]. Whereas CD4 Treg lymphocytes play the opposite role, i.e., inhibit self-reactive cytotoxic T and Th17 lymphocytes, which initiate self-reactive cytotoxic T lymphocyte activation [53]. Self-reactive T lymphocytes are known to avoid clonal deletion or differentiation into the thymic Treg cell lineage early in life and enter the pancreatic lymph nodes [54]. To manipulate the cytokine axis to the equilibrated Treg/Th17 axis, cytokines, recombinant proteins, or antibodies are used. After antigen presentation, IL-2 triggers CD4 Treg lymphocytes; the dosage of recombinant IL-2 is the major one responsible for mimicking the Treg/Th17 pathway homeostasis [55,56].
Further possible antibodies for T1D therapy are anti-IL1 and anti-tumor necrosis factor-alpha (TNF-α), signaling molecules involved in inflammation and then destruction of pancreatic β-cells. Anti-TNF medications have previously been used to treat chronic pro-inflammatory autoimmune diseases, such as RA [57,58]. Despite the fact that etanercept (anti-TNFα) that binds soluble TNF was unable to prevent the development or progression of T1D [59]. Antibodies against TNF, Etanercept 24 weeks treatment, may benefit T1D since clinical studies have shown that this significant molecule decreases in HbA1c, and improves β-cell preservation compared to the placebo group [38].
There are increases in clinical trials that show a renewed interest in soluble cytokines, anti-cytokines recombinant monoclonal antibodies, or the corresponding cytokine receptors that are more often used [60]. The human monoclonal antibodies against TNF, such as adalimumab and etanercept, show the binding affinity towards the soluble TNFα than its TNFα receptors [61]. The TNFα binding with adalimumab (anti-TNFα] induces a conformational change that trimerizes TNFα receptors on Treg lymphocytes, which later causes their expansion [62]. The anti-IL17 recombinant monoclonal antibody Brodalumab, which suppresses the Th17 pathway, has been approved for a few autoimmune diseases [63]. While preclinical studies conducted in animal models suggest that the Th17 pathway is also associated with autoimmune T1D [64], clinical trials are necessary to reveal whether antibodies that suppress Th17 signaling have therapeutic effects on T1D. Based on a clinical study with an anti-IL21 drug that affects T lymphocyte activation. IL-21 is mainly secreted by CD4 helper T lymphocytes. IL-23 is another proinflammatory cytokine directed in immune intervention studies because shares the homology with p40 subunit with IL-12, another proinflammatory cytokine. Both IL-12 and IL-23 are involved in amplifying proinflammatory pathways and thus play critical roles in autoimmune processes. Ustekinumab is a mAb that deals with the homology/shared p40 subunit of IL-12 and IL-23, thereby blocking subsequent signaling and differentiation of central immune pathways. It intensifies the cytotoxic activity of natural killer cells [65]. Later, there were two clinical trials conducted, the first one based on IL1 antagonists Canakinumab (anti-IL1 mAb) [41] and second, Anakinra (IL1R antagonist) [66], both unsuccessful in showing any statistically significant differences compared to placebo control group. This suggests that targeting single cytokine inhibition will likely not cure or provide a beneficial effect in the treatment of autoimmune T1D, due to the redundancy of pro-inflammatory cytokines associated in the onset and maintenance of the disease.
Suppressing B Cells: Despite the fact that autoimmune T1D is a T lymphocyte-mediated autoimmune disease, B lymphocytes also play a pathogenic role in antigen-presenting cells that modulate the pancreatic microenvironment [67]. Researchers linked B lymphocytes to the pathogenesis of autoimmune T1D via antigen presentation and T cell activation. CD20, a cell surface protein expressed on B lymphocytes, is required for B cell activation and proliferation and has emerged as a reliable therapeutic target with rituximab (anti-CD20). The TrialNet study group looked at how a short sequence of four infusions spread out over 30 days could delay the drop in C peptide by more than eight months in patients with newly diagnosed T1D. Nonetheless, the decline in C-peptide and β-cell function was essentially the same as the placebo control after two years [33].
Dendritic Cells based tolerogenic approach: The potent antigen-presenting and linker cells of innate and adaptive lymphocytes, i.e., dendritic cells (DCs) are generated from bone marrow’s progenitor cells, which can maintain peripheral tolerance. The likelihood of creating tolerogenic DCs opens new therapeutic tactics in the prevention or remission of autoimmunity. DCs are the edge between innate and adaptive immune responses and are crucial for the initiation of antibody-humoral and cellular immunity [68], including T lymphocyte differentiation and activation [69]. They also support maintaining immunological tolerance by clearing apoptotic cells rapidly in the body. To govern if this DC can prevent autoimmune T1D by re-establishing peripheral immunological tolerance while using the primed DCs with dead cells from pancreatic β-cells, Marrin-Gallen et al. initially showed a lower disease incidence than the control group when NOD mice were injected with DCs primed with nitrilase-1 apoptotic bodies [70]. However, disappointing results were achieved in humans with T1D [71]. Later, a randomized, double-blind, phase I study was conducted with a few n = 10 subjects with T1D. These individuals were injected with ten million DCs into the abdomen intradermally once every 2 weeks for a total of four administrations and supervised for a year showed that a treatment regimen with autologous DCs, in a naive state or directed ex vivo to a tolerogenic immunosuppressive condition or state, is safe and well-tolerated. The DCs elevated the frequency of a potentially advantageous B220+CD11c− B-cell population, at least in T1D autoimmunity [72]. However, several other human clinical studies resulted in disappointing results, which tells us that multiple agents contribute to failure compared to the success seen in the mice study. Similarly, this autoimmune T1D is associated with differential exposure to environmental factors [71].
Therapies that induce immune-suppressing T-cells: Pancreatic β-cells can be shielded from the autoimmune responses via the presence of Treg cells, which are immune-suppressive. Various strategies were investigated to expand the pancreas’ Treg cells, including anti-CD3 mAb and IL-2 (a signaling molecule called interleukin-2).
Vaccines: In general, autoimmune diseases are characterized by the overproduction of specific cytokines, which primarily disrupt the immune system. As a result, many studies have focused on inhibiting excessive cytokine production to halt these autoimmune diseases. As a result, vaccine development is desirable in order to stimulate the immune system to neutralize the specific protein produced in excess. Tolerance can reverse pancreatic β-cell destruction if attacking T-cells expose a small number of specific proteins (or parts of proteins called peptides) from cells. C19-A2 proinsulin, a peptide, has been shown to regulate the immune response elicited by antigen-specific T lymphocytes.
Furthermore, this peptide was found to improve pancreatic β-cell function in T1D patients. In adults, a clinical trial using intramuscular DNA vaccinated with a proinsulin-encoding plasmid (BHT-3021), which demonstrated overall safety, suggests that this strategy may have the potential to be used for treatment in the future. They discovered that antigen-specific CD8+ T lymphocytes and preserved C-peptide levels were lower in the periphery while on this treatment. The primary immune cells found in the pancreas after infiltration include autoreactive B and T cells, natural killer (NK) cells, macrophages, and DCs [73]. The increased levels of IFN-I found in T1D patients’ pancreatic islets and peripheral blood may contribute to disease progression; thus, blocking or downregulating this cytokine may aid in the prevention of autoimmune T1D pathogenesis [74]. Neovacs initiated IFNα-kinoid vaccine-based treatment approach for T1D.
Antigen-Specific Immunomodulatory Approaches: Several autoimmune diseases, antigen-specific therapeutic approaches have been studied [75], which modulate the inactivation of autoreactive T cells in T1D [76]. A list of antigen-specific immunomodulatory advances is shown in Figure 2 and Table 2.
Immune checkpoint-based therapy: Latest innovations focus on the considerable influence of inhibitory receptors (IRs) based therapy, facilitating peripheral tolerance in autoimmune diseases. The depletion or inhibition of specific blockade studies in animals, humans, and their association with inspiring disease outcomes highlight the prospective clinical benefits of enhancing IR signaling (agonism), specifically immune-checkpoint targets like CTLA4, PD1, TIM3, LAG3, and TIGIT to treat autoimmune diseases other than cancers [121]. A growing understanding of the significant role of IRs in cancer and recent developments in autoimmunity suggests that IR agonism may help prevent and manage autoimmune diseases, as reviewed in a review article by Stephanie Grebinoski and Dario AA Vignali [122].
Immune cell therapy: In autoimmune T1D, immune system cells mistakenly identify insulin hormone-producing pancreatic β-cells in pancreatic islets as foreign and dangerous; the primary goal of immune cell-based therapy is to disrupt these autoimmune signaling events. However, in the early stages of development, immune cell-based therapy is one of the most important ideas for developing a cure for autoimmune T1D. Replacing the missing insulin-producing β-cells may be able to restore average insulin production and cure T1D patients. However, initial attempts to transplant pancreatic cells have largely failed, owing to immune reactions. Another limitation is the scarcity of HLA-specific donors.
One promising approach is epitope-specific intervention, which can suppress the autoimmune response while avoiding the side effects of other therapies. Even though autoimmune T1D is polyclonal, with multiple T lymphocyte epitopes and pancreatic β-cell-specific autoantibodies, single epitope-specific medications can suppress the polyclonal immune response and reverse T1D in preclinical models [123]. The administration of specific immunomodulatory cells may aid in the reduction of the aggressive immune response against pancreatic β-cells. Tolerogenic DCs are known to suppress immune responses through a variety of mechanisms. Infusion of these cells, along with Treg cells, is being investigated as a potential strategy to prevent or reduce pancreatic β-cell destruction by the immune system. There are a few GMP-compliant procedures for isolating autologous CD4 Treg lymphocytes from peripheral blood [124]. The adaptive immune cell transfer of autologous CD4 Treg lymphocytes triggers a tolerogenic state [125], and few clinical trials are ongoing to determine whether it can efficaciously ameliorate autoimmunity. Few other new approaches develop Treg cells or cytotoxic T cells expressing specific receptors that enable the cells to target the pancreatic β-cells, increasing the therapy’s effectiveness while reducing the cells’ unwanted effects on other non-target regions of the body. In conclusion, a current roadblock to development in the cell therapy field is the lack of an efficient system to generate the “precise” auto or islet-Ag-specific cytotoxic CD4, CD8, or Treg lymphocytes that can be used for immune cell-based therapies in autoimmune diabetes or other diseases, as shown in Figure 3.
Tolerogenic autologous DC was generated in vivo and infused intradermally into a T1D patient. Later, in a clinical trial, they discovered that those patients tolerated the therapy well. Still, more research is needed to determine whether they can change the course of the disease [72]. Current exercise to find islet-specific T cell receptors and vital epitopes in autoimmune T1D, which may also allow for the generation of antigen-specific Treg or T lymphocytes in vitro using gene-editing tools [126].
Imcyse, a Belgian biotech company, develops imotopes, which generate pancreatic β-cell antigen-specific cytolytic memory CD4+ lymphocytes while precisely eliminating antigen-presenting cells (APCs) presenting the same T-cell epitope through apoptosis. The pathogenic T lymphocytes activated by other epitopes on the same APC are also destroyed by the cytolytic memory CD4+ lymphocytes (bystander effect). Imotope technology will now be able to precisely eliminate pathogenic auto-immune responses and cure autoimmune T1D. The lead molecule, IMCY-0098TM, is an imotope that has been developed to treat T1D and is currently in clinical trial 2b (NCT04524949). Imcyse’s strategy, when used consistently, aids in the prevention and treatment of diseases for which there are no current therapeutic options, as well as the potential cure of patients without impairing immune defense.
Overcoming immunotherapy challenges: While various immunotherapies are being tested in clinical trials for the treatment of autoimmune T1D, several issues must be addressed. The factors involved in T1D differ among individuals with this condition, complicating treatment because a single type of treatment or therapy may not be sufficient for all patients. For example, not all T1D patients or individuals have islet cell inflammation. As a result, treatments aimed at reducing inflammation in islet cells may be ineffective in patients who do not have this type of inflammation. Given the individual differences in T1D characteristics, personalized medicine for individuals may be a highly beneficial approach in the coming year.
A new facet of T1D immunotherapy is achieving satisfactory efficacy. Triggering or targeting immune cells may result in a wide range of responses, reducing treatment efficacy and causing adverse effects. Physicians and researchers are attempting to address this issue through a variety of therapeutic approaches. Combining therapies that act through different mechanisms of action improves treatment efficacy. Varieties must be chosen in such a way that efficacy is improved, and side effects are minimized.
3. Conclusions and Future Perspectives
However, there is still a long way to go; new autoimmune diseases, particularly T1D immunotherapy, hold great promise for T1D patients. In the future, we may even be able to use single or multi-immunotherapy to prevent the condition before it manifests. This novel strategy is possible because we now have the technological capability to identify individuals at risk of T1D before significant damage to the pancreatic β-cells occurs. The past 30 years have taught us that immunotherapy has the potential to treat or preserve autoimmune T1D. However, we have yet to see a breakthrough because we have yet to crack the code in terms of an acceptable safety/efficacy balance for treating autoimmune T1D. Immune cell-based approaches to T1D treatment may offer the possibility of lifelong insulin administration in people with autoimmune T1D. Assume we used T1D patients in the early stages of the disease, when β-cell mass destruction is low or a significant percentage of β-cell mass remains. The in vivo expansion of pancreatic islet antigen-specific Treg therapy via adoptive transfer will be beneficial. In this case, attacking CD8 T lymphocytes and administering ex vivo prepared autologous tolerogenic DCs are promising immune cell-based therapies that may effectively preserve pancreatic β-cell mass and function. Since the discovery of insulin by Banting and Best in 1922, several recent developments have investigated autoimmune T1D management. In the near future, new skills will allow us to treat autoimmune diseases as well as prevent or reverse them.
There are still a few unanswered questions. Even though significant progress in our understanding of the immunobiology of autoimmune T1D allows for the development of novel therapeutics that may be beneficial and clinically safe for preserving autoimmune T1D.
Key questions that must be answered:
- What factors/agents cause the initial destruction of pancreatic β-cells in T1D patients?
- ○
- Genetic or epigenetic modifications;
- ○
- Viral infection;
- ○
- Gut microbial flora;
- ○
- Environmental or diet and nutrition;
- ○
- Aging or developmental changes destroy β-cells.
- Once pancreatic cells are destroyed, how do immune cells select their cognitive epitope; why do these epitopes express or present more? What factor or cells cause them?
- Self-epitope/antigen enters circulation—how/why do CD4/CD8 T cells begin recognizing self-antigen, indicating that TCR rearrangement occurred previously with the corresponding epitope? What causes the immune tolerance system to fail to circulate these immune cells?
- Once in the circulation, β-cells antigen/markers, specific T cells are generated; how do these T cells begin attacking remaining β-cell masses who give them command? Is there any expression of pancreas-specific signals or chemokine receptors on these cells? How they infiltrated the pancreatic islet to destroy-cells.
- What causes Treg cells to become inactive?
- How can we prevent an unwelcome immune attack on pancreatic β-cells? The best strategy is to hide and attack.
- Hide: Modify pancreatic β-cell-specific antigen recognition/presentation to preserve β-cell mass.
- Attack: Attacking the rebel immune cells is another option; we can generate antigen-specific Treg or cytotoxic CD4/CD8 T cells to kill the rebel immune cells.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All figures created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Firestein, G.S.; Budd, R.C.; Harris, E.D.; McInnes, I.B.; Sergent, J.S. Kelley’s Textbook of Rheumatology, 8th ed.; WB Saunders: Philadephia, PA, USA, 2008. [Google Scholar]
- Goldman, L.; Ausiello, D.A. Cecil Medicine; Saunders Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- Herold, K.C.; Vignali, D.A.A.; Cooke, A.; Bluestone, J.A. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol. 2013, 13, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Tisch, R.; McDevitt, H. Insulin-dependent diabetes mellitus. Cell 1996, 85, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.S.; Bluestone, J.A. THE NOD MOUSE: A Model of Immune Dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485. [Google Scholar] [CrossRef] [PubMed]
- Burn, P. Type 1 diabetes. Nat. Rev. Drug Discov. 2010, 9, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Głowińska-Olszewska, B.; Szabłowski, M.; Panas, P.; Żołądek, K.; Jamiołkowska-Sztabkowska, M.; Milewska, A.J.; Kadłubiska, A.; Polkowska, A.; Łuczyński, W.; Bossowski, A. Increasing Co-occurrence of Additional Autoimmune Disorders at Diabetes Type 1 Onset Among Children and Adolescents Diagnosed in Years 2010–2018—Single-Center Study. Front. Endocrinol. 2020, 11, 476. [Google Scholar] [CrossRef]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef] [Green Version]
- Van Belle, T.L.; Coppieters, K.T.; Von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Atkinson, M.A. The Pathogenesis and Natural History of Type 1 Diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007641. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- Pepper, A.R.; Bruni, A.; Shapiro, A.J. Clinical islet transplantation: Is the future finally now? Curr. Opin. Organ Transplant. 2018, 23, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Bassi, R.; Fiorina, P. Impact of Islet Transplantation on Diabetes Complications and Quality of Life. Curr. Diabetes Rep. 2011, 11, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, Å. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Harjutsalo, V.; Podar, T.; Tuomilehto, J. Cumulative Incidence of Type 1 Diabetes in 10,168 Siblings of Finnish Young-Onset Type 1 Diabetic Patients. Diabetes 2005, 54, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for Islet Autoimmunity among Monozygotic Twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Liu, Y.-F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Skyler, J.S.; Pugliese, A. Immunotherapy Trials for Type 1 Diabetes: The Contribution of George Eisenbarth. Diabetes Technol. Ther. 2013, 15, S2-13–S2-30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feutren, G.; Assan, R.; Karsenty, G.; Du Rostu, H.; Sirmai, J.; Papoz, L.; Vialettes, B.; Vexiau, P.; Rodier, M.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset: Results of a multicentre double-blind trial. Lancet 1986, 328, 119–124. [Google Scholar] [CrossRef]
- Sigal, N.H.; Dumont, F.J.; Cyclosporin, A. FK-506, and rapamycin: Pharmacologic probes of lymphocyte signal transduction. Annu. Rev. Immunol. 1992, 10, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Canadian-European Randomized Control Trial Group. Cyclosporin-induced remission of IDDM after early intervention: Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. Diabetes 1988, 37, 1574–1582. [Google Scholar] [CrossRef]
- Füchtenbusch, M.; Kredel, K.; Bonifacio, E.; Schnell, O.; Ziegler, A.G. Exposure to exogenous insulin promotes IgG1 and the T-helper 2-associated IgG4 responses to insulin but not to other islet autoantigens. Diabetes 2000, 49, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural basis of assembly of the human T cell receptor–CD3 complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Long, S.A.; Thorpe, J.; DeBerg, H.A.; Gersuk, V.; Eddy, J.A.; Harris, K.M.; Ehlers, M.; Herold, K.C.; Nepom, G.T.; Linsley, P.S. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016, 1, eaai7793. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Anti-CD3 drug keeps diabetes at bay. Nat. Biotechnol. 2019, 37, 1099–1101. [Google Scholar] [CrossRef]
- Wang, X.; Ni, L.; Chang, D.; Lu, H.; Jiang, Y.; Kim, B.-S.; Wang, A.; Liu, X.; Zhong, B.; Yang, X.; et al. Cyclic AMP-Responsive Element-Binding Protein (CREB) is Critical in Autoimmunity by Promoting Th17 but Inhibiting Treg Cell Differentiation. EBioMedicine 2017, 25, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, L.; Sheng, X.; Chen, W.; Tang, H.; Sheng, H.; Xi, B.; Zang, Y.Q. T-cell vaccination leads to suppression of intrapancreatic Th17 cells through Stat3-mediated RORγt inhibition in autoimmune diabetes. Cell Res. 2011, 21, 1358–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stifter, K.; Schuster, C.; Schlosser, M.; Boehm, B.O.; Schirmbeck, R. Exploring the induction of preproinsulin-specific Foxp3+ CD4+ Treg cells that inhibit CD8+ T cell-mediated autoimmune diabetes by DNA vaccination. Sci. Rep. 2016, 6, 29419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, L.; Robey, E.A.; Hsieh, C.-S. Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 2019, 19, 7–18. [Google Scholar] [CrossRef]
- Kitashima, D.Y.; Kobayashi, T.; Woodring, T.; Idouchi, K.; Döbel, T.; Voisin, B.; Adachi, T.; Ouchi, T.; Takahashi, H.; Nishifuji, K.; et al. Langerhans Cells Prevent Autoimmunity via Expansion of Keratinocyte Antigen-Specific Regulatory T Cells. EBioMedicine 2018, 27, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Boardman, D.; Levings, M.K. Cancer immunotherapies repurposed for use in autoimmunity. Nat. Biomed. Eng. 2019, 3, 259–263. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Tack, C.J.; Kleijwegt, F.S.; Van Riel, P.L.C.M.; Roep, B.O. Development of type 1 diabetes in a patient treated with anti-TNF-α therapy for active rheumatoid arthritis. Diabetologia 2009, 52, 1442–1444. [Google Scholar] [CrossRef] [Green Version]
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept Treatment in Children With New-Onset Type 1 Diabetes: Pilot Randomized, Placebo-Controlled, Double-Blind Study: Response to Peters. Diabetes Care 2009, 32, e154. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Faustman, D.L. TNF, TNF inducers, and TNFR2 agonists: A new path to type 1 diabetes treatment. Diabetes/Metab. Res. Rev. 2018, 34, e2941. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.A.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.; Bakery, H.H.; Allam, G. The potential pathogenic role of IL-17/Th17 cells in both type 1 and type 2 diabetes mellitus. Biomed. Pharmacother. 2018, 101, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Nambam, B.; Haller, M.J. Updates on Immune Therapies in Type 1 Diabetes. Eur. Endocrinol. 2016, 12, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, S.M.; Wang, X.; Chen, Y.-G.; Jia, S.; Kaldunski, M.; Greenbaum, C.J.; Mandrup-Poulsen, T.; Hessner, M.J.; the Type 1 Diabetes TrialNet Canakinumab Study Group; the AIDA Study Group. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur. J. Immunol. 2015, 46, 1030–1046. [Google Scholar] [CrossRef] [Green Version]
- Van Asseldonk, E.J.; Van Poppel, P.C.; Ballak, D.B.; Stienstra, R.; Netea, M.G.; Tack, C.J. One week treatment with the IL-1 receptor antagonist anakinra leads to a sustained improvement in insulin sensitivity in insulin resistant patients with type 1 diabetes mellitus. Clin. Immunol. 2015, 160, 155–162. [Google Scholar] [CrossRef]
- Mariño, E.; Silveira, P.A.; Stolp, J.; Grey, S.T. B cell-directed therapies in type 1 diabetes. Trends Immunol. 2011, 32, 287–294. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Bundy, B.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; Moran, A.; Raskin, P.; et al. B-Lymphocyte Depletion With Rituximab and β-Cell Function: Two-Year Results. Diabetes Care 2014, 37, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Dakic, A. Development of dendritic cell system. Cell. Mol. Immunol. 2004, 1, 112–118. [Google Scholar]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic cells based immunotherapy. Am. J. Cancer Res. 2017, 7, 2091. [Google Scholar]
- Marin-Gallen, S.; Clemente-Casares, X.; Planas, R.; Pujol-Autonell, I.; Carrascal, J.; Carrillo, J.; Ampudia, R.; Verdaguer, J.; Pujol-Borrell, R.; Borràs, F.E.; et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clin. Exp. Immunol. 2009, 160, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Creusot, R.J.; Giannoukakis, N.; Trucco, M.; Clare-Salzler, M.J.; Fathman, C.G. It’s Time to Bring Dendritic Cell Therapy to Type 1 Diabetes. Diabetes 2013, 63, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (Safety) Study of Autologous Tolerogenic Dendritic Cells in Type 1 Diabetic Patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regnell, S.E.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaisar, N.; Jurczyk, A.; Wang, J.P. Potential role of type I interferon in the pathogenic process leading to type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Harrison, L.C.; von Herrath, M.G. Trials in type 1 diabetes: Antigen-specific therapies. Clin. Immunol. 2013, 149, 345–355. [Google Scholar] [CrossRef]
- Luo, X.; Herold, K.C.; Miller, S.D. Immunotherapy of Type 1 Diabetes: Where Are We and Where Should We Be Going? Immunity 2010, 32, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.; Abbas, A.K. The enemy within: Keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2002, 2, 11–19. [Google Scholar] [CrossRef]
- Grebinoski, S.; Vignali, D.A. Inhibitory receptor agonists: The future of autoimmune disease therapeutics? Curr. Opin. Immunol. 2020, 67, 1–9. [Google Scholar] [CrossRef]
- Tang, Q.; Henriksen, K.J.; Bi, M.; Finger, E.B.; Szot, G.; Ye, J.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In Vitro–expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T. Illuminating immune privilege—A role for regulatory T cells in preventing rejection. N. Engl. J. Med. 2011, 365, 956–957. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Falzetti, F.; Carotti, A.; Terenzi, A.; Pierini, A.; Massei, M.S.; Amico, L.; Urbani, E.; et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 2014, 124, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I. Generation of human islet-specific regulatory T cells by TCR gene transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safari, F.; Farajnia, S.; Arya, M.; Zarredar, H.; Nasrolahi, A. CRISPR and personalized Treg therapy: New insights into the treatment of rheumatoid arthritis. Immunopharmacol. Immunotoxicol. 2018, 40, 201–211. [Google Scholar] [CrossRef]
- Aronson, R.; Gottlieb, P.A.; Christiansen, J.S.; Donner, T.W.; Bosi, E.; Bode, B.W.; Pozzilli, P.; the DEFEND Investigator Group. Low-Dose Otelixizumab Anti-CD3 Monoclonal Antibody DEFEND-1 Study: Results of the Randomized Phase III Study in Recent-Onset Human Type 1 Diabetes. Diabetes Care 2014, 37, 2746–2754. [Google Scholar] [CrossRef] [Green Version]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J.; Bode, B.; Aronoff, S.; Holand, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Hagopian, W.; Ferry, R.J.; Sherry, N.; Carlin, D.; Bonvini, E.; Johnson, S.; Stein, K.; Koenig, S.; Daifotis, A.; Herold, K.; et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: Two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 2013, 62, 3901–3908. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef] [Green Version]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Costimulation Modulation With Abatacept in Patients With Recent-Onset Type 1 Diabetes: Follow-up 1 Year After Cessation of Treatment. Diabetes Care 2014, 37, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Rigby, M.R.; Harris, K.M.; Pinckney, A.; DiMeglio, L.A.; Rendell, M.S.; Felner, E.I.; Dostou, J.M.; Gitelman, S.E.; Griffin, K.J.; Tsalikian, E.; et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J. Clin. Investig. 2015, 125, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Moran, A.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; Raskin, P.; et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 2013, 381, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Sumpter, K.M.; Adhikari, S.; Grishman, E.K.; White, P.C. Preliminary studies related to anti-interleukin-1β therapy in children with newly diagnosed type 1 diabetes. Pediatr. Diabetes 2011, 12, 656–667. [Google Scholar] [CrossRef]
- Bonifacio, E.; Ziegler, A.G.; Klingensmith, G.; Schober, E.; Bingley, P.J.; Rottenkolber, M.; Theil, A.; Eugster, A.; Puff, R.; Peplow, C.; et al. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: The Pre-POINT randomized clinical trial. JAMA 2015, 313, 1541–1549. [Google Scholar] [CrossRef]
- Voltarelli, J.C.; Couri, C.E.B.; Stracieri, A.B.P.L.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.R.; Foss-Freitas, M.C.; Simões, B.P.; et al. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in Newly Diagnosed Type 1 Diabetes Mellitus. JAMA J. Am. Med. Assoc. 2007, 297, 1568–1576. [Google Scholar] [CrossRef] [Green Version]
- D’Addio, F.; Vasquez, A.V.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation in New-Onset Type 1 Diabetes: A Multicenter Analysis. Diabetes 2014, 63, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.; Rosenthal, S.M.; Shuster, J.J.; Zou, B.; Brusko, T.M.; Hulme, M.A.; Wasserfall, C.; et al. Anti-thymocyte globulin/G-CSF treatment preserves β cell function in patients with established type 1 diabetes. J. Clin. Investig. 2015, 125, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.W.; Landry, L.G.; McDaniel, K.A.; Yu, L.; Campbell-Thompson, M.; Kwok, W.W.; Jones, K.L.; Gottlieb, P.A.; Kappler, J.W.; Tang, Q.; et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes 2016, 66, 722–734. [Google Scholar] [CrossRef] [Green Version]
- Wong, F.S.; Karttunen, J.; Dumont, C.; Wen, L.; Visintin, I.; Pilip, I.M.; Shastri, N.; Pamer, E.G.; Janeway, C.A. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 1999, 5, 1026–1031. [Google Scholar] [CrossRef]
- Chen, J.; Grieshaber, S.; Mathews, C.E. Methods to Assess Beta Cell Death Mediated by Cytotoxic T Lymphocytes. J. Vis. Exp. 2011, 2011, e2724. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, B.; Dudek, N.L.; McKenzie, M.D.; Purcell, A.; Brooks, A.; Gellert, S.; Colman, P.G.; Harrison, L.C.; Lew, A.; Thomas, H.E.; et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J. Clin. Investig. 2006, 116, 3258–3265. [Google Scholar] [CrossRef]
- Nakayama, M.; Abiru, N.; Moriyama, H.; Babaya, N.; Liu, E.; Miao, D.; Yu, L.; Wegmann, D.R.; Hutton, J.C.; Elliott, J.F.; et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005, 435, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Kohm, A.P.; McMahon, J.S.; Luo, X.; Miller, S.D. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J. Autoimmun. 2012, 39, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Nakayama, M.; Eisenbarth, G.S. Insulin as an autoantigen in NOD/human diabetes. Curr. Opin. Immunol. 2008, 20, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Kracht, M.J.L.; Van Lummel, M.; Nikolic, T.; Joosten, A.M.; Laban, S.; van der Slik, A.; van Veelen, P.; Carlotti, F.; De Koning, F.C.E.J.P.; Hoeben, R.; et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat. Med. 2017, 23, 501–507. [Google Scholar] [CrossRef]
- Jin, N.; Wang, Y.; Crawford, F.; White, J.; Marrack, P.; Dai, S.; Kappler, J.W. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, 13318–13323. [Google Scholar] [CrossRef] [Green Version]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016, 351, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Wiles, T.A.; Powell, R.; Michel, C.R.; Beard, K.S.; Hohenstein, A.; Bradley, B.; Reisdorph, N.; Haskins, K.; Delong, T. Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J. Proteome Res. 2019, 18, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, D.L.; Clare-Salzler, M.; Tian, J.; Forsthuber, T.; Ting, G.S.P.; Robinson, P.; Atkinson, M.A.; Sercarz, E.E.; Tobin, A.J.; Lehmann, P.V. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature 1993, 366, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Kaufman, D.L.; Newman, D.; Tobin, A.J.; Maclaren, N.K. Islet cell cytoplasmic autoantibody reactivity to glutamate decarboxylase in insulin-dependent diabetes. J. Clin. Investig. 1993, 91, 350–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloot, N.C.; Daniel, D.; Norbury-Glaser, M.; Wegmann, D.R. Peripheral T cell Clones from NOD Mice Specific for GAD65 Peptides: Lack of Islet Responsiveness or Diabetogenicity. J. Autoimmun. 1996, 9, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Videbæk, N.; Harach, S.; Phillips, J.; Hutchings, P.; Ozegbe, P.; Michelsen, B.K.; Cooke, A. An islet-homing NOD CD8+cytotoxic T cell clone recognizes GAD65and causes insulitis. J. Autoimmun. 2003, 20, 97–109. [Google Scholar] [CrossRef]
- Wenzlau, J.M.; Walter, M.; Gardner, T.J.; Frisch, L.M.; Yu, L.; Eisenbarth, G.S.; Ziegler, A.-G.; Davidson, H.W.; Hutton, J.C. Kinetics of the Post-Onset Decline in Zinc Transporter 8 Autoantibodies in Type 1 Diabetic Human Subjects. J. Clin. Endocrinol. Metab. 2010, 95, 4712–4719. [Google Scholar] [CrossRef] [Green Version]
- Dang, M.; Rockell, J.; Wagner, R.; Wenzlau, J.M.; Yu, L.; Hutton, J.C.; Gottlieb, P.A.; Davidson, H.W. Human Type 1 Diabetes Is Associated with T Cell Autoimmunity to Zinc Transporter. J. Immunol. 2011, 186, 6056–6063. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Calderon, B.; Vomund, A.N.; Unanue, E.R. ZnT8-Reactive T Cells Are Weakly Pathogenic in NOD Mice but Can Participate in Diabetes Under Inflammatory Conditions. Diabetes 2014, 63, 3438–3448. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Gu, Y.; Bian, L.; Shi, Y.; Cai, Y.; Chen, Y.; Chen, H.; Qian, L.; Wu, X.; Xu, K.; et al. Characterization of immune response to novel HLA-A2-restricted epitopes from zinc transporter 8 in type 1 diabetes. Vaccine 2015, 34, 854–862. [Google Scholar] [CrossRef]
- Émmanuelle, É.; Kratzer, R.; Arnoux, J.-B.; Barilleau, E.; Hamel, Y.; Marchi, C.; Beltrand, J.; Michaud, B.; Chatenoud, L.; Robert, J.-J.; et al. ZnT8 Is a Major CD8+ T Cell–Recognized Autoantigen in Pediatric Type 1 Diabetes. Diabetes 2012, 61, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Wenzlau, J.M.; Frisch, L.M.; Hutton, J.C.; Davidson, H.W. Mapping of conformational autoantibody epitopes in ZNT8. Diabetes/Metab. Res. Rev. 2011, 27, 883–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubosaki, A.; Miura, J.; Notkins, A.L. IA-2 is not required for the development of diabetes in NOD mice. Diabetologia 2004, 47, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Q.; Standifer, N.E.; Qin, H.; Gottlieb, P.; Verchere, C.B.; Nepom, G.T.; Tan, R.; Panagiotopoulos, C. Recognition of HLA class I-restricted beta-cell epitopes in type 1 diabetes. Diabetes 2006, 55, 3068–3074. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Miao, D.; Michels, A.; Steck, A.; Dong, F.; Rewers, M.; Yu, L. A multiplex assay combining insulin, GAD, IA-2 and transglutaminase autoantibodies to facilitate screening for pre-type 1 diabetes and celiac disease. J. Immunol. Methods 2016, 430, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, E.; Hutton, J.C.; Eisenbarth, G.S. Molecular Cloning and Characterization of the Human Transmembrane Protein Tyrosine Phosphatase Homologue, Phogrin, an Autoantigen of Type 1 Diabetes. Biochem. Biophys. Res. Commun. 1996, 227, 440–447. [Google Scholar] [CrossRef]
- Kawasaki, E.; Eisenbarth, G.S.; Wasmeier, C.; Hutton, J.C. Autoantibodies to protein tyrosine phosphatase-like proteins in type I diabetes: Overlapping specificities to phogrin and ICA512/IA-2. Diabetes 1996, 45, 1344–1349. [Google Scholar] [CrossRef]
- Kelemen, K.; Crawford, M.L.; Gill, R.G.; Hutton, J.C.; Wegmann, D. Cellular immune response to phogrin in the NOD mouse: Cloned T-cells cause destruction of islet transplants. Diabetes 1999, 48, 1529–1534. [Google Scholar] [CrossRef]
- Kelemen, K.; Gottlieb, P.A.; Putnam, A.L.; Davidson, H.W.; Wegmann, D.R.; Hutton, J.C. HLA-DQ8-associated T cell responses to the diabetes autoantigen phogrin (IA-2 beta) in human prediabetes. J. Immunol. 2004, 172, 3955–3962. [Google Scholar] [CrossRef] [Green Version]
- Karges, W.; Hammond-McKibben, D.; Gaedigk, R.; Shibuya, N.; Cheung, R.; Dosch, H.M. Loss of self-tolerance to ICA69 in nonobese diabetic mice. Diabetes 1997, 46, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Astsaturov, I.; Gaedigk, R.; Hammond-McKibben, D.; Pilon, M.; Song, A.; Kubiak, V.; Karges, W.; Arpaia, E.; McKerlie, C.; et al. ICA69null Nonobese Diabetic Mice Develop Diabetes, but Resist Disease Acceleration by Cyclophosphamide. J. Immunol. 2002, 168, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, S.; Tsui, H.; Lau, A.; Song, A.; Li, X.; Cheung, R.K.; Sampson, A.; Afifiyan, F.; Elford, A.; Jackowski, G.; et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat. Med. 2003, 9, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bergerot, I.; Elliott, J.F.; Harrison, L.C.; Abiru, N.; Eisenbarth, G.S.; Delovitch, T.L. Evidence that a peptide spanning the B-C junction of proinsulin is an early Autoantigen epitope in the pathogenesis of type 1 diabetes. J. Immunol. 2001, 167, 4926–4935. [Google Scholar] [CrossRef] [Green Version]
- Spitzenberger, F.; Pietropaolo, S.; Verkade, P.; Habermann, B.; Lacas-Gervais, S.; Mziaut, H.; Pietropaolo, M.; Solimena, M. Islet Cell Autoantigen of 69 kDa Is an Arfaptin-related Protein Associated with the Golgi Complex of Insulinoma INS-1 Cells. J. Biol. Chem. 2003, 278, 26166–26173. [Google Scholar] [CrossRef] [Green Version]
- Stadinski, B.D.; Delong, T.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Piganelli, J.D.; Barbour, G.; Bradley, B.; Crawford, F.; et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 2010, 11, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, P.A.; Delong, T.; Baker, R.L.; Fitzgerald-Miller, L.; Wagner, R.; Cook, G.; Rewers, M.R.; Michels, A.; Haskins, K. Chromogranin A is a T cell antigen in human type 1 diabetes. J. Autoimmun. 2014, 50, 38–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoopour, E.; Krougly, O.; Lee-Chan, E.; Haeryfar, S.M.; Singh, B. Detection of vasostatin-1-specific CD8+ T cells in non-obese diabetic mice that contribute to diabetes pathogenesis. Clin. Exp. Immunol. 2016, 185, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Delong, T.; Baker, R.L.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Barbour, G.; Bradley, B.; Haskins, K. Islet Amyloid Polypeptide Is a Target Antigen for Diabetogenic CD4+ T Cells. Diabetes 2011, 60, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.L.; Delong, T.; Barbour, G.; Bradley, B.; Nakayama, M.; Haskins, K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J. Immunol. 2013, 191, 3990–3994. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Mahiddine, K.; Baker, R.L.; Haskins, K.; Guerder, S. The T Cell Repertoire–Diversifying Enzyme TSSP Contributes to Thymic Selection of Diabetogenic CD4 T Cell Specificities Reactive to ChgA and IAPP Autoantigens. J. Immunol. 2015, 195, 1964–1973. [Google Scholar] [CrossRef] [Green Version]
- Wiles, T.A.; Delong, T.; Baker, R.; Bradley, B.; Barbour, G.; Powell, R.L.; Reisdorph, N.; Haskins, K. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J. Autoimmun. 2017, 78, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Serra, P.; Amrani, A.; Yamanouchi, J.; Marée, A.F.M.; Edelstein-Keshet, L.; Santamaria, P. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: High efficiency of a low-affinity peptide. Nat. Med. 2005, 11, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Danke, N.A.; Berger, D.; Reichstetter, S.; Reijonen, H.; Greenbaum, C.; Pihoker, C.; James, E.A.; Kwok, W.W. Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein-Reactive CD4+ T Cells in Human Subjects. J. Immunol. 2006, 176, 2781–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.-J.; Chee, J.; Sutherland, R.M.; Thomas, H.E.; Zhan, Y.; Krishnamurthy, B.; Kay, T.W.H.; Lew, A.M. Functional cytotoxic T lymphocytes against IGRP 206-214 predict diabetes in the non-obese diabetic mouse. Immunol. Cell Biol. 2014, 92, 640–644. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Type I Diabetes immune response.

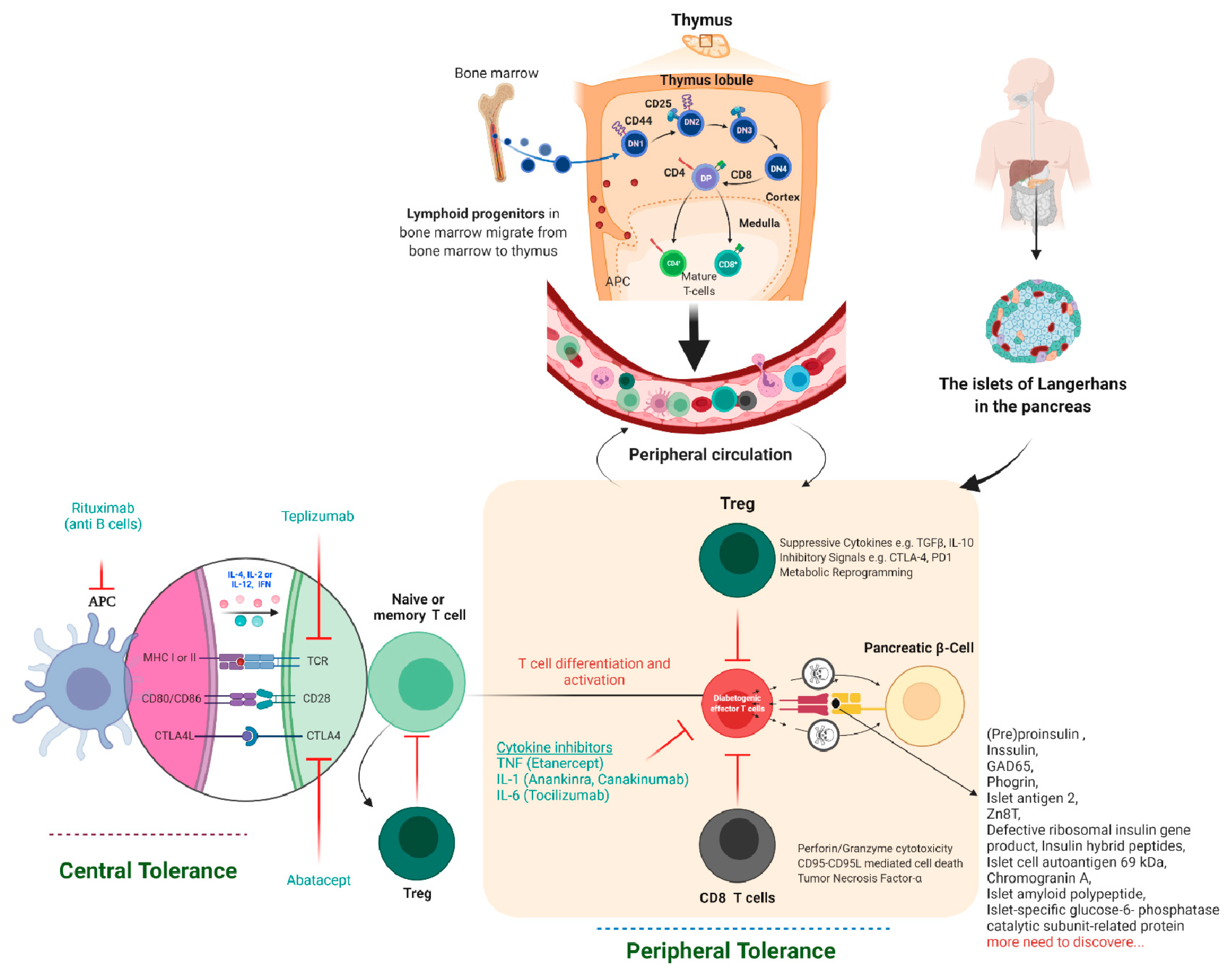
Figure 2.
Immune tolerance, therapeutics, and pancreatic β-cell-specific antigen discovered till now in T1D. The lymphoid progenitor cells are initially generated in the host bone marrow by special hematopoietic stem cells. The T lymphocytes travel to the thymus, where the thymus-based central tolerance mechanisms train T cells to discriminate between self and non-self (adverse selection). CD4 regulatory T cells (Tregs) and diabetogenic T lymphocytes may identify self or pancreatic β-cell-specific antigens, but at different affinities, which might explain their destructive actions β-cell. Subsequently, live T lymphocytes arrive in the blood and lymph nodes’ peripheral circulatory system and clash with their specific peptide-MHC/HLA complex. Specifically, in autoimmune T1D, these T lymphocytes are specific for pancreatic β-cell proteins such as insulin, GAD55, or so many others discovered to date. If these pancreatic β-cell-specific T lymphocytes and their respective antigen/epitopes are displayed by the MHC/HLA of antigen-presenting cells (DC, Macrophage, or B cells), T lymphocytes will become initiated in the lymph node, migrate to the pancreatic islets, and begin the demolition of β-cell in an antigen-specific manner. Tregs characterize the suppressive lymphocytes mainly responsible for peripheral immunological tolerance and try to inhibit these events. If the host body is unable to stop the autoimmune attack on pancreatic β-cells, insulin deficiency, hyperglycemia, and, eventually, autoimmune T1D will result. The majority of signaling events occur in the peripheral-local environment, in the lymph nodes and pancreas, and cannot be tracked using biomarkers.
Figure 2.
Immune tolerance, therapeutics, and pancreatic β-cell-specific antigen discovered till now in T1D. The lymphoid progenitor cells are initially generated in the host bone marrow by special hematopoietic stem cells. The T lymphocytes travel to the thymus, where the thymus-based central tolerance mechanisms train T cells to discriminate between self and non-self (adverse selection). CD4 regulatory T cells (Tregs) and diabetogenic T lymphocytes may identify self or pancreatic β-cell-specific antigens, but at different affinities, which might explain their destructive actions β-cell. Subsequently, live T lymphocytes arrive in the blood and lymph nodes’ peripheral circulatory system and clash with their specific peptide-MHC/HLA complex. Specifically, in autoimmune T1D, these T lymphocytes are specific for pancreatic β-cell proteins such as insulin, GAD55, or so many others discovered to date. If these pancreatic β-cell-specific T lymphocytes and their respective antigen/epitopes are displayed by the MHC/HLA of antigen-presenting cells (DC, Macrophage, or B cells), T lymphocytes will become initiated in the lymph node, migrate to the pancreatic islets, and begin the demolition of β-cell in an antigen-specific manner. Tregs characterize the suppressive lymphocytes mainly responsible for peripheral immunological tolerance and try to inhibit these events. If the host body is unable to stop the autoimmune attack on pancreatic β-cells, insulin deficiency, hyperglycemia, and, eventually, autoimmune T1D will result. The majority of signaling events occur in the peripheral-local environment, in the lymph nodes and pancreas, and cannot be tracked using biomarkers.

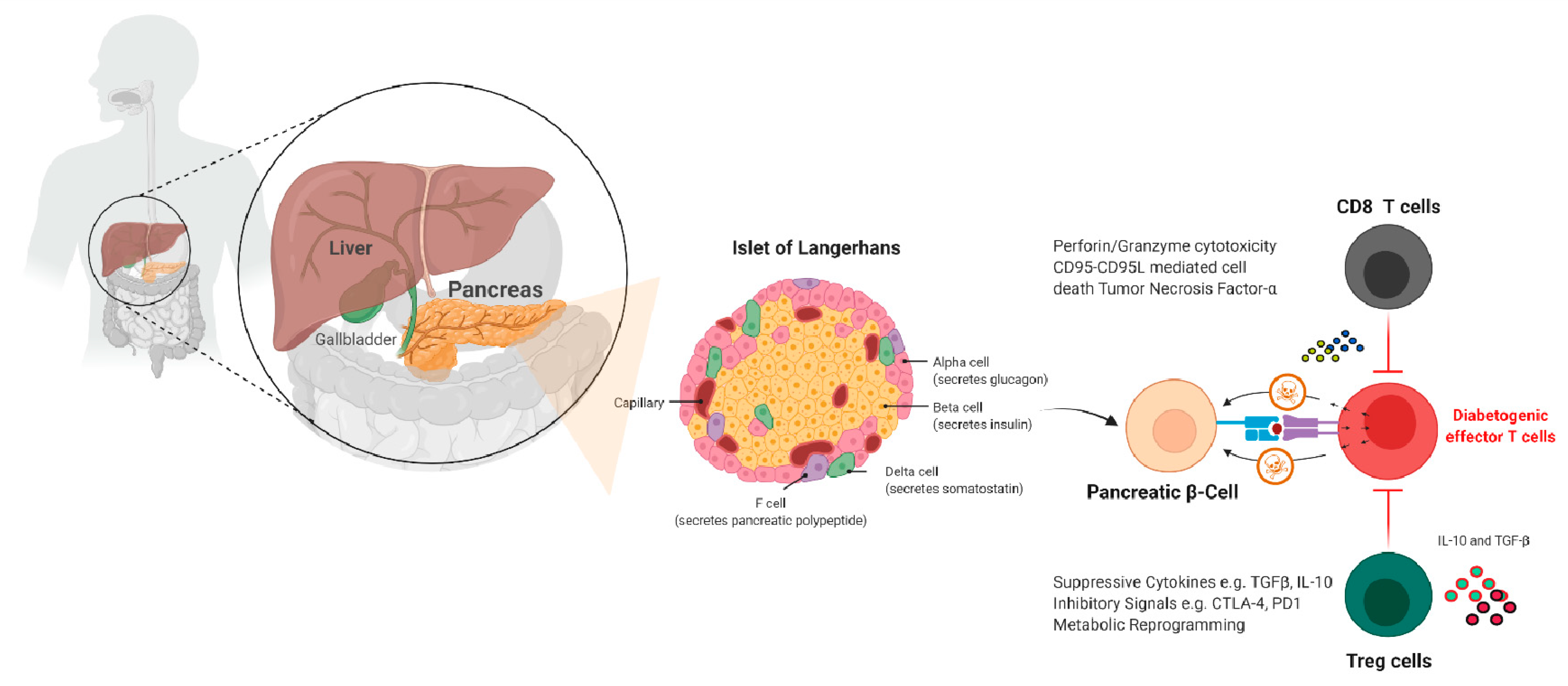
Figure 3.
Pathogenesis of T1D and immunotherapy. This diagram depicts pancreatic islets and the various pancreatic cells found within them, such as delta, alpha, F, Acinus, and our target β-cell. Once a pancreatic β-cell is destroyed, its peptides are presented by immune cells, and the immune cell infiltrates the pancreas and destroys the remaining β-cells. We can protect these pancreatic-cells that secrete insulin by hiding or destroying diabetogenic effector T cells by generating β-cells that are epitope/antigen-specific cytotoxic CD4 or CD8 T cells or Treg cells that kill.
Figure 3.
Pathogenesis of T1D and immunotherapy. This diagram depicts pancreatic islets and the various pancreatic cells found within them, such as delta, alpha, F, Acinus, and our target β-cell. Once a pancreatic β-cell is destroyed, its peptides are presented by immune cells, and the immune cell infiltrates the pancreas and destroys the remaining β-cells. We can protect these pancreatic-cells that secrete insulin by hiding or destroying diabetogenic effector T cells by generating β-cells that are epitope/antigen-specific cytotoxic CD4 or CD8 T cells or Treg cells that kill.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative Immune-based therapeutic intervention study in autoimmune T1D and outcomes.
| Therapeutic Agents | Study/Authors and Intervention | Outcome | Citations |
|---|---|---|---|
T cell-based:
| DEFEND-1, 2 (Otelexizumab) | There was no EBV in the therapy group, but there was no statistically significant difference in 2-h MMTT AUC C-peptide at 12 months. | [28] |
| Protégé (Teplizumab) | At 1 year, there was no significant difference in HbA1c1 < 6.5 percent or insulin dose < 0.5 U/kg per day: At year 2, AUC C-peptide in the high dose group was considerably greater than in the placebo group. | [29,30] | |
| AbATE (Teplizumab) | The treatment group’s baseline adjusted AUC C-peptide reduced at year 2 was considerably lower. | [31] | |
| B cell-based: The monoclonal anti-CD20 antibody, which blocks the B cell function | Rituximab | HbA1c lowers as the rate of C peptide declines and insulin levels decrease. | [32,33] |
| Co-stimulation blockade | TrialNet CTLA4-Ig (abatacept); CTLA-4-IgG1 chimeric protein acts as a decoy receptor for CD80/86 and blocks CD28-CD80/86 induced co-stimulation of T-cells | Significantly higher stimulated C-peptide 2-h AUC in the treated group at the end of treatment and 1-year post-treatment | [34,35] |
| TIDAL (alafacept); Alafacept: chimeric protein (2 LFA-3 molecule-IgG1) binds to CD2 and blocks T-cell-stimulation | Significantly higher stimulated AUC C-peptide in the treatment group compared to placebo; insulin use lower in the treatment group | [36] | |
| Cytokine-based: IL-2 agonist | Aldesleukin; IL-2 maintains Treg population and function | A dose-dependent elevation of Treg cells in the treatment group compared to placebo | [37] |
| TNF antagonism | Etanercept | HbA1c decreases while endogenous insulin production increases. | [38] |
| IL-1 receptor blockade | Anakinra |
| [39,40] |
| IL-1beta antagonism | Canakinumab | There was no C peptide reaction | [39] |
| IL-1 receptor blockade IL-1beta antagonism | Anakinra/canakinumab | Immunomodulation/reverse relationship between inflammation and C peptide stimulation | [41] |
| Anti-IL-6 therapy | Tocilizumab in New-onset T1D (EXTEND) | Ongoing study | Clinical trial NCT02293837 |
| Antigen-based therapy: | Antigen-specific therapies may involve direct targeting of pathogenic T cells and/or boosting Tregs for bystander suppression | Tregs were shown to be more prevalent in those who got a larger dose of oral insulin (62.5 mg) | [42] |
| Treg-based: | Expansion of autologous Treg cells | A subset of adoptively transferred Treg is still in circulation (25% of peak) at year 1, with no significant adverse effects. C-peptide preservation in those receiving a lower dose | [20] |
| DC-based: | In T1D individuals who get their autologous DCs exhibited limited output. In this study, autologous DCs were given by infused via abdominal intradermal injections each 2 weeks apart | The autologous DC-based therapy was very well tolerated; no important differences were seen in glycemia | [37] |
| Combination therapy |
| C-peptide significantly increased at 30 months follow up; increased side effects | [43] |
| 32% were insulin-free at 4 years, maintenance of C-peptide, but with increased side effects | [44] | ||
| Mean AUC C-peptide at 12 months was significantly higher in the study group compared to the placebo group | Low-dose ATG + plus pegylated G-CSF [45] |
AbATE = Autoimmunity Blocking Antibody for Tolerance in Recently Diagnosed T1Ds, ATG = anti-thymocyte globulin, AUC = area under curve, CTLA-4 = cytotoxic T-lymphocyte-associated antigen, DEFEND = Durable Response Therapy Evaluation for Early or T1D = Type 1 Diabetes, EBV = Epstein Barr virus, G-CSF = granulocyte colony-stimulating factor, HbA1c = glycated haemoglobin, IL-2 = interleukin-2, LFA-3 = leukocyte function antigen-3, MMTT = mixed meal tolerance test, Pre-POINT = Primary intervention with Oral Insulin for Prevention of T1D in infants at high genetic risk, TCR = T-cell receptor, Teffs = T effector cells, TIDAL = Type 1 Diabetes with Alefacept, Tregs = T regulatory cells, DC = dendritic cells.
Table 2.
Autoantigen secreted by pancreatic β-cells in NOD mouse or T1D patients.
| T1D Autoantigens | Tissue Distribution | Source (NOD Mouse or T1D Patients) | Effector CD4 and/or CD8 T Cells | Citations |
|---|---|---|---|---|
| (Pre) proinsulin | β cells, thymus | Mouse and human | CD4 and CD8 | [77,78,79,80] |
| Insulin | Islet cells | Mouse and human | CD4 and CD8 | [78,81,82,83] |
| A defective ribosomal insulin gene product | Islet cells | Human | CD8 | [84] |
| Hybrid insulin peptides (HIPs) | Islet cells | Mouse and human | CD4 | [85,86,87,88] |
| Glutamic acid decarboxylase (GAD65) | Islet cells, adrenal gland, CNS, neurons, testis, ovary | Mouse and human | CD4 and CD8 | [89,90,91,92] |
| Zinc transporter 8 (ZnT8) | Pancreatic β cells | Mouse and human | CD4 and CD8 | [93,94,95,96,97,98] |
| Tyrosine phosphatase like autoantigen or insulinoma antigen-2 (IA-2; ICA512, PTPRN) | Islets | Human | CD4 and CD8 | [99,100,101] |
| IA-2β (Phogrin, PTPRN2) | Islets | Mouse and human | CD4 | [102,103,104,105] |
| Islet cell autoantigen of 69 kDa (ICA69) | Pancreas, heart, and brain | Human | CD4 | [106,107,108,109,110] |
| Chromogranin A | Neuroendocrine cells | Mouse and human | CD4 and CD8 | [111,112,113] |
| Islet amyloid polypeptide (ppIAPP) | Islets | Mouse and human | CD4 and CD8 | [114,115,116,117] |
| IGRP; islet-specific glucose-6-phosphatase catalytic subunit-related protein | Islets | Mouse and human | CD4 and CD8 | [80,118,119,120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rathod, S. Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes. Diabetology 2022, 3, 79-96. https://doi.org/10.3390/diabetology3010007
AMA Style
Rathod S. Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes. Diabetology. 2022; 3(1):79-96. https://doi.org/10.3390/diabetology3010007
Chicago/Turabian StyleRathod, Sanjay. 2022. "Novel Insights into the Immunotherapy-Based Treatment Strategy for Autoimmune Type 1 Diabetes" Diabetology 3, no. 1: 79-96. https://doi.org/10.3390/diabetology3010007