
Analysis of Hydrogen in Inorganic Materials and Coatings: A Critical Review


Institute of Physics of the Czech Academy of Sciences, Na Slovance 2, 182 21 Praha 8, Czech Republic
Hydrogen 2021, 2(2), 225-245; https://doi.org/10.3390/hydrogen2020012
Submission received: 10 April 2021
/
Revised: 7 May 2021
/
Accepted: 12 May 2021
/
Published: 4 June 2021
Abstract
:The currently used bulk analysis and depth profiling methods for hydrogen in inorganic materials and inorganic coatings are reviewed. Bulk analysis of hydrogen is based on fusion of macroscopic samples in an inert gas and the detection of the thereby released gaseous H2 using inert gas fusion (IGF) and thermal desorption spectroscopy (TDS). They offer excellent accuracy and sensitivity. Depth profiling methods involve glow discharge optical emission spectroscopy and mass spectrometry (GDOES and GDMS), laser-induced breakdown spectroscopy (LIBS), secondary ion mass spectrometry (SIMS), nuclear reaction analysis (NRA), and elastic recoil detection analysis (ERDA). The principles of all these methods are explained in terms of the methodology, calibration procedures, analytical performance, and major application areas. The synergies and the complementarity of various methods of hydrogen analysis are described. The existing literature about these methods is critically evaluated, and major papers concerning each method are listed.
1. Introduction
The presence of hydrogen in various materials and the ways in which it affects their properties has been the subject of attention of the scientific community for decades. A major area developed in this context since the first half of 20th century was hydrogen in metals [1,2]. Other application areas gradually followed, reflecting the progress in materials technology ever since. The analysis of hydrogen is special in many respects, and it makes sense to treat it separately from other elements. In this context, it is worth mentioning the electron structure of the hydrogen atom; hydrogen has only one electron and does not possess deep inner shells. Therefore, many common methods of material characterization, such as X-ray fluorescence analysis, electron microanalysis (EDX/WDX systems), electron spectroscopies (Auger electron spectroscopy, X-ray photoelectron spectroscopy), and some other cannot be used for hydrogen, for fundamental reasons. Methods for hydrogen analysis are diverse, in every respect, and information about them is scattered through the literature, ranging from analytical chemistry to metallurgy, nuclear science, and other application areas. The aim of this review is to categorize these methods and describe their principles and analytical performance, so that the reader may understand how they work, the kind of information they can provide, and their figures of merit, as well as get an idea of how to address the specific analytical tasks related to hydrogen arising in various areas of science and technology.
Analytical tasks involving hydrogen analysis can be divided into two large groups: (1) bulk analysis concerns the volume of the sample, i.e., where the concentration of hydrogen in the sample as a whole is of interest, and (2) depth profile analysis (depth profiling) in which the distribution of hydrogen is measured as function of the depth beneath the sample surface. It concerns various thin film systems, materials with a modified composition close to the surface, e.g., materials modified by various chemical treatments, ion implantation, etc. This very basic division of the methods of hydrogen analysis is reflected in the structure of this review.
2. Bulk Analysis of Hydrogen
The earliest analyses of hydrogen concerned metallurgical applications. This is also the area in which the most robust methods of hydrogen analysis emerged. In ferrous metallurgy, hydrogen in steels is an important issue, in connection with unwanted effects such as hydrogen embrittlement [3,4,5], hydrogen flaking [6], and problems caused by hydrogen in welding [7,8,9]. Another area worth mentioning is electrochemical charging of metals by hydrogen and cathodic protection against it [10]. In nonferrous metallurgy, issues related to hydrogen are important in nickel alloys [11], aluminum [12,13], titanium [14], and magnesium [15], as reviewed, e.g., in [16]. Another metal extensively studied in connection with hydrogen is palladium [17] and some other precious metals, e.g., in the role of hydrogen-storage materials. In the production and processing of engineering metals and alloys, it is usually desirable to minimize the amount of hydrogen present in the product.
Typical ranges of hydrogen concentration to be analyzed in metallurgical applications can be assessed on the basis of the plots in Figure 1. The solubility of hydrogen in various metals depends on temperature and crystallographic structure; for example, in austenite (γ-Fe, fcc lattice), it is higher than in ferrite (α-Fe or δ-Fe, bcc lattice). In ferrous metallurgy, welding, casting, etc., it is common to distinguish between “diffusible” and “residual” hydrogen [8,9], whereby the former can be released by diffusion at room or only slightly elevated temperature, unlike the latter. Residual hydrogen concentration depends, in addition to the solubility in the melt, on the conditions of solidification and/or the subsequent heat treatment of the material under study. The content of hydrogen in a material is expressed in various units: atomic or weight fraction (percentage) or the volume of gaseous molecular hydrogen in certain amount of the material, assuming atmospheric pressure and room temperature. These are the ordinate units in Figure 1. This unit, y = cm3 H2 at 0.1 MPa per 100 g of material, is linked to the weight percentage of hydrogen in the material, cH, by Avogadro’s law.
It is worth noting that weight concentrations of hydrogen are often lower than atomic concentrations by 1–2 orders of magnitude in number, because the atomic mass of hydrogen, Mr(H) = 1, is low compared to other elements (e.g., Mr(Fe) ≈ 56). This needs to be considered when comparing the sensitivity of analytical methods for hydrogen and other elements, as sensitivity is usually expressed in weight units.
Another issue, relevant to the analysis of hydrogen, is transport properties; hydrogen exhibits a high diffusion coefficient in some materials. Therefore, sample preparation and handling may be critical. In many cases, samples need to be kept at a low temperature before analysis (liquid nitrogen), to prevent losses by diffusion to the sample surface. The diffusion coefficient of hydrogen interstitials in bcc iron metal (α-Fe) is D ≈ 7 × 10−5 cm2·s−1 at room temperature (25 °C), which yields a characteristic diffusion length d of ~0.2 mm (d2 = 6 Dt) within t = 1 s [3]. On the other hand, in austenite (γ-Fe, fcc lattice), diffusion of hydrogen is slower by orders of magnitude, and austenitic stainless steels are even used as stable reference materials for hydrogen analysis, with an indefinite shelf-life. As an example, the grade 1.4546.9 steel with 0.97 ± 0.05 ppm H (Stahl-H1 by BAM Berlin) can be permanently stored at 4 °C [18].
2.1. Inert Gas Fusion
Inert gas fusion, (IGF) [20], is a classical method of hydrogen analysis, supported by commercial instruments from several manufacturers. It has become the industry standard in this area and a benchmark for other methods. Sometimes, it is also called gas fusion analysis (GFA); both names are used. Experimental setup is relatively simple; it does not require high vacuum or expensive mass spectrometric detection systems. The sample is placed in a flow of inert gas, typically argon, at atmospheric pressure, and hydrogen present in the sample is released in the form of H2 gas by heating the sample, typically high above the melting point. The amount of hydrogen in the (Ar + H2) gas flow after the furnace is measured by a thermal conductivity (TC) detector [21,22], as the presence of hydrogen in argon substantially enhances the thermal conductivity of the gas. Alternatively, hydrogen in the (Ar + H2) mixture after the furnace can be oxidized to water vapor, H2O, and detected by an infrared absorption detector (IR) [23]; it is based upon the absorption of infrared radiation as it passes through a volume of gas. The advantage of IR detectors is that, by placing a suitable filter into the beam of originally broadband radiation, it is possible to achieve absorption in a narrow region around a specific wavelength. Therefore, the detector can be made selective to a certain molecular gas, such as H2O, without sensing other molecules. Other gases of interest in the IGF analysis also have strong absorption bands in the IR region, such as carbon monoxide, CO, and carbon dioxide, CO2, and can, thus, be detected by IR detectors. Unlike IR detectors, TC detectors are not selective; every component of the gas mixture contributes to its thermal conductivity. Consequently, it is necessary to modify the potentially complex gas mixture originating from the sample so that the TC detector is reached by a simple binary mixture, i.e., the carrier gas + the gas to be measured. In addition to hydrogen, there are other elements in the sample causing the release of gases, typically oxygen and nitrogen. In the IGF method, the sample is melted in a graphite crucible, heated by a high current passing through it, at a power of several kilowatts. Oxygen from the sample reacts with the graphite to form CO and, in some cases, a mixture of CO and CO2. Nitrogen from the sample is released as N2 gas. To facilitate fusion of refractory metals, a flux is usually added to the sample, either pure nickel or pure tin.
The characteristics of the detectors described above and the nature of the gases potentially released from the sample imply different possible configurations of an IGF analyzer, with the potential to analyze H, O, and N simultaneously or in various combinations. Two such configurations, relevant to the analysis of hydrogen, are presented in Figure 2. The reactions utilized to modify the composition of the gas on its path from the furnace to the detector and the corresponding reagents are in Table 1. In the analysis with TC detection (Figure 2a), the gas mixture entering the detector may contain nitrogen in addition to hydrogen. To enable the analysis of this mixture based on thermal conductivity, a column with a molecular sieve is put before the detector. Hydrogen passes through the column much more quickly than nitrogen (similarly to gas chromatography), and it is, thus, possible to separate H2 from N2 using a suitable time window in which the TC signal is collected.
Detection limits are in the sub-ppm range, based on 1 g sample mass, i.e., in the sub-μg range of the absolute amount of hydrogen. The analysis is relative, based on calibration with appropriate reference materials. Alternatively, calibration by known additions of H2 gas into the carrier gas is also possible. This makes IGF a primary reference method for hydrogen analysis. The analyses are metrologically traceable to fundamental measurement units, in a very wide range of concentrations: from sub-ppm up to the concentrations in hydrides, typically several weight percent of H. IGF analyses are typically performed on dedicated instruments and are considered routine. The typical duration of an IGF analysis is a few minutes.
2.2. Thermal Desorption Analysis
Thermal desorption analysis (TDA) or thermal desorption spectrometry (TDS) [20,24,25,26] is closely related to IGF and also uses thermal decomposition of the sample. However, instead of a rapid heating above the melting point, the sample is heated slowly, and the rate at which hydrogen is released is registered as a function of temperature. Typical temperature ramp rates are 1–10 °C/min, while the temperature range usually does not exceed 1000 °C. Quartz tube furnaces are used most frequently, with nitrogen as the carrier gas and TC detection. This experimental setup is also called hot extraction. The total amount of hydrogen released throughout the analysis or in a selected temperature region is obtained by integrating the desorption curve. To increase sensitivity, a quadrupole mass spectrometer may be used as a detector (thermal desorption mass spectrometry, TDMS). Typical detection limits based on 1 g sample mass are, thus, in the sub-ppb range (~0.1 ng H) [27].
Applications involve the analysis of “trapped” and “diffusible” hydrogen in steels [24,27,28,29], hydrogen trapping [25] (see Figure 3), hydrogen-induced second phases, and hydrogen embrittlement mechanisms in Ti alloys [14,24] and hydrogen-storage materials [30]. TDS analysis, together with thermogravimetry, was also used to characterize Ti–H reference materials for the analysis of higher concentrations of hydrogen [31]. For completeness, it is also worth mentioning here the more traditional low-temperature volumetric method of the determination of diffusible hydrogen by its collection at room or slightly elevated temperature under mercury [8,9] or glycerine.
3. Analysis of Hydrogen in Coatings and Thin Films
The analysis of hydrogen in coatings can be divided into two categories: (1) analysis of total amount of hydrogen in a coating and (2) a depth-resolved analysis (depth profiling). The latter case is relevant especially if the depth distribution of hydrogen is uneven. The total amount of hydrogen can then be established by the integration of the depth profile (see Section 3.1.1, Equations (4) and (5)). In principle, for the former task, the methods described in Section 2 can be used, provided that the amount of hydrogen in the coating is high enough and if the contribution of hydrogen present in the substrate is negligible or can be subtracted, on the basis of another analysis, of the plain substrate, without the coating (a “blank” measurement). As an illustration, a 1 μm thick TiH2 layer on a 0.2 mm thick substrate (e.g., stainless steel) can be considered. The hydrogen concentration in the coating is ~4% (in mass units) and the density is ρ(TiH2) = 3.76 g·cm−3 [32], which corresponds to an amount of hydrogen in the coating of 15 μg·cm−2. For the IGF or TDS analysis, it is feasible to consider a relatively large sample, up to say 10 cm2, which, for a one-side coverage by the coating, means 150 μg of hydrogen. This is more than by two orders of magnitude more than the detection limit of IGF or TDS; hence, it is high enough for accurate analysis (see Section 2.1 and Section 2.2). To keep the amount of hydrogen in the substrate comparable (same) with that in the coating, the hydrogen concentration in the substrate should be (4/200) × ρ(TiH2)/ρ(steel) ≈ 0.0095% = 95 ppm, while ρ(steel) = 7.9 g·cm−3. Hydrogen concentrations in steel around 100 ppm are much higher than what can be expected, provided that the process of making the coating does not itself add a large amount of hydrogen into the substrate. Hence, analyses like this are feasible. If the sensitivity for hydrogen is enhanced by mass spectrometric detection (TDMS), this approach should also work for coatings with a much lower hydrogen concentration than that in TiH2. TDMS analyses of titanium deuteride films on a quartz substrate were described in [33]. The size of the sample may also play a role; for example, a 10 cm2 sample may seem too large to be sacrificed for a single analysis. An advantage of this is, however, that the IGF and TDA methods are accurate and “absolute”, in the sense mentioned in Section 2. This does not necessarily apply to some other methods listed below.
Another fact worth mentioning is that the most common methods of elemental analysis in laboratories involved in materials science are those implemented in electron microscopes: electron microanalysis (EDS/WDS) and electron energy loss spectroscopy (EELS). As neither of these can be used to analyze hydrogen, and because, in many labs, no direct methods of hydrogen analysis are available, the effects of hydrogen are sometimes treated indirectly, without actual analysis. Such an approach was used, e.g., for hydrogen in boron-doped diamond-like carbon layers, in which hydrogen in the coating was sensed by a peak at 188 eV in the X-ray photoelectron spectrum (XPS) of the 1s level of boron, indicating the presence of B–H bonds [34]. Although information about the B–H bonds in the spectrum exists, this can hardly be regarded as a true analysis of hydrogen.
3.1. Emission Spectroscopy and Mass Spectrometry
The two emission spectroscopic methods listed below, GDOES (glow discharge optical emission spectroscopy) and LIBS (laser-induced breakdown spectroscopy), are well established for general analytical purposes and are quite common in laboratories involved in solid-state analytical chemistry. The measurements themselves are relatively simple; however, the subsequent interpretation of the data, to establish the concentration of hydrogen, may not be easy, particularly in complex matrices. The emission spectrum of atomic hydrogen has been known for more than a century and was the central point in the development of the quantum theory, during the first third of the 20th century. The following are the lines of choice for the analysis of hydrogen: λ = 121.57 nm (Lyman α) and three lines of the Balmer series, λ = 656.27 nm (Hα), λ = 486.13 nm (Hβ), and λ = 434.05 nm (Hγ). The line at 121.57 nm is the most sensitive one but requires a spectrometer designed for the vacuum UV region, as this radiation is strongly absorbed by atmospheric oxygen. Such instruments are common in GDOES, not so much in LIBS. The optical system has to be either evacuated or purged with an inert gas. The second most sensitive hydrogen line is Hα, λ = 656.27 nm, in the visible region (red), and it is used in both LIBS and GDOES.
Related to GDOES is glow discharge mass spectrometry (GDMS). It also uses a glow discharge atomization and ionization source, attached to a mass spectrometer, and the analysis is based on the signal of selected ions (see below). Another mass-spectrometric method is secondary ion mass spectrometry (SIMS), in which the ions of the analyzed material are not created in a glow discharge plasma but by bombarding the surface under study using a focused ion beam, under high-vacuum conditions. GDMS and SIMS are more expensive techniques than GDOES and are also less common.
3.1.1. Glow Discharge Optical Emission Spectroscopy (GDOES) and Glow Discharge Mass Spectrometry (GDMS)
In GDOES and GDMS [35,36], the sample acts as cathode in a glow discharge (GD) in argon and is atomized by cathodic sputtering. The sputtered atoms are excited and ionized in the GD plasma and emit characteristic radiation that is analyzed by an optical spectrometer (GDOES). In GDMS, the ions are extracted from the plasma and analyzed by a mass spectrometer. The sample is atomized continuously, layer-by-layer, as the erosion crater proceeds into the depth, and this makes both methods suitable for depth profiling, provided that the signal is registered as function of time. Total depths that can be analyzed range from tens of nanometers to >100 μm. Typical sputtering rates for metallic matrices are ~10–100 nm/s and the total duration of the analysis depends on the thickness of the layer under study. Nonmetallic matrices such as nitrides, oxides, and carbides have typically lower sputtering rates than metals. Depth resolution is depth-dependent, typically at ~10% of the depth below the original sample surface. GDOES and GDMS depth profiles, not quantified and involving hydrogen, are shown in Figure 4a [37] and Figure 5. A sufficiently high sampling rate, necessary for GDMS depth profiling of several elements in very thin layers, is possible by using a time-of-flight (TOF) mass spectrometer [38].
A crucial point in the analysis by GDOES and GDMS is quantification, i.e., the conversion of raw data (qualitative depth profiles of signal intensity versus time of sputtering) into the composition-versus-depth (quantitative) profiles. Both GDOES and GDMS are used as relative methods; quantification is based on empirical a priori information acquired by analyzing suitable reference samples. Various calibration models and quantification schemes have been developed for both GDOES [35,39] and GDMS [40,41].
In GDOES, the approximation of matrix-independent emission yields (MIEY) is widely used, under which the intensity IE,λ of an emission line λ of an element E is proportional to its concentration cE,M in the matrix M analyzed, as well as to the sputtering rate qM of that matrix.
where RE,λ is a constant called the emission yield, expressing the relative sensitivity of the line λ in the specific instrument and under the specific discharge conditions used. Emission yields are treated as constants, independent of the matrix analyzed. Equation (2) shows that the quantities controlling the spectral response are interlinked, as the sputtering rate qM depends on the matrix (see the plot in Figure 6). Hence, in a qualitative profile, a higher intensity of the line is not necessarily caused by a higher concentration of the respective element. The abscissa of the profile may also be distorted; the time needed to pass different layers is not only proportional to their thickness, but also inversely proportional to their sputtering rates. The effects of a variable sputtering rate may be very substantial; for example, the sputtering rate of diamond-like carbon is ~30 times lower than that of iron [42]. This calibration model, Equation (2), allows for accurate quantitative multi-matrix analyses, provided that suitable reference materials with a known composition and known sputter rates are available. Another prerequisite is that virtually all elements present at significant concentrations are measured. The achievable accuracy is well comparable with best methods routinely used for bulk compositional analysis (e.g. X-ray fluorescence, XRF). The presence of hydrogen, however, adds to the complexity, as hydrogen affects GD excitation and ionization of other elements. Such effects were first described in chromium [43] and have been extensively studied in both GDOES [44,45] and GDMS [46]. Various correction schemes have been proposed to handle such situations, e.g., in the analysis of N-implanted Ti [47]. Accurate quantitative analysis of hydrogen by GDOES usually requires the calibration and quantification to be matrix-matched and supported by suitable reference materials. They include bulk standards, such as a hot-pressed powder of TiH2 mixed with Cu [48] and hydrogenated Zr–Nb alloy [49], or various coatings, e.g., electrodeposited Zn(H), magnetron-sputtered WC(H), or amorphous hydrogenated silicon, a-Si:H, produced by chemical vapor deposition [37,50].
If there is some hydrogen in the coating but not in the substrate, the GDOES analysis can be verified and linked to an “absolute” method, such as IGF or TDS, as follows: quantification of an unknown sample, based on Equation (2), results in the first step in the calculated concentrations of the elements present, cE,M(t), and calculated sputtering rate, qM(t), as functions of time, t. The conversion of the time coordinate t into depth x, x = x(t), is based on the definition of the sputtering rate (the sputtered mass-per-time).
where S is the area of the erosion crater, and ρM(x) is the density at the depth x. The depth coordinate x can then be obtained by integration of Equation (3), provided that the density ρM is known as function of the composition. There is, however, no universal formula expressing the density of a material as function of its composition; hence, the resulting profile as function of x is only approximate. The total mass of hydrogen in the sputtered area of the coating, mH, is
By substituting from Equation (3), this can be rewritten as
i.e., the integration over depth x can be replaced by integration over time, while the density is implicitly included in the sputtering rate qM(t). Equation (5) gives, therefore, an accurate correspondence between the measured depth profile and the total amount of hydrogen mH. mH can then be compared with the amount of hydrogen as obtained by IGF or TDS.
GDOES is a rapid and affordable method, suitable for obtaining basic semiquantitative information about hydrogen in a coating or for comparison of hydrogen depth profiles in a series of similar samples that differ, e.g., by the conditions of preparation. A flat sample is needed, with an area exceeding the dimensions of the sealing o-ring of the GD source (typical diameter of several mm). The results of GDOES can eventually be further refined by another method or by comparison with GDOES data from matrix-specific standards, created specifically for the application under study. GDOES analyses of hydrogen described in the literature include, in addition to the already mentioned applications, hydrogen in various thin-film and multilayer systems [51], e.g., in diamond-like carbon films [42,52]. The former paper is also a comparison with ERDA (elastic recoil detection analysis; see Section 3.2.1). Other applications involve Si/C anodes of Li-ion batteries [53], analysis of deuterium in W [54] and Ti, and research into plasma-facing materials for thermonuclear fusion [55]. In that study, integrated GDOES depth profiles were compared with analyses by TDS, as mentioned above. Compared to GDOES, much fewer analyses of hydrogen have been reported using GDMS. They include, e.g., analysis of H in gold layers [56] or a-Si:H solar cells [38].
3.1.2. Laser-Induced Breakdown Spectrometry (LIBS)
In laser-induced breakdown spectrometry (LIBS) [57], the sample is irradiated by a laser shot which ablates a small amount of sample material, generating a plasma plume. Its light emission is collected and analyzed by an optical spectrometer. Unlike GDOES, the small diameter of the focused laser beam enables spatially resolved analysis [58]. However, the performance of LIBS in accurate multi-element and multi-matrix analyses is inferior to that of GDOES. A simple but still accurate semi-empirical calibration model, similar to the MIEY approximation in GDOES, does not exist in LIBS, because of the much more complex physics involved in the atomization (ablation) and excitation processes. There have been attempts to interpret the LIBS spectra on the basis of the concepts of fundamental plasma physics, using plasma parameters such as excitation temperatures (calibration-free (CF) LIBS [59,60,61]). Their analytical outcome, however, has so far been rather semi-quantitative, with few exceptions, such as the analysis of glass [62]. Best results can be achieved with matrix-specific calibrations of hydrogen in simple matrices (e.g., a homogeneous matrix consisting of two or few elements only). This is how most of the hydrogen analyses mentioned below were done. A rudimentary depth profiling by LIBS is possible through successive analyses at the same spot [63]; however, the depth range is limited compared to GDOES at both ends, both for thin and thick coatings. Furthermore, only a coarse depth sampling is possible, as the depth step is equal to the depth of the ablation crater resulting from a single laser shot.
Applications of the analysis of hydrogen by LIBS involve hydrogen and deuterium analysis in plasma-facing materials for thermonuclear fusion devices [60,61,63,64], materials for nuclear fission reactors, such as Zircalloy [65], hydrogen in weldments [66], hydrogen-storage materials (MgH2) [67], isotopic analysis of hydrogen in titanium [68,69], and even hydrogen analysis in H-bearing minerals on Mars by the rover Curiosity [70]. Isotopic analysis is traditionally a domain of mass spectrometry; however, the isotopic shift in the hydrogen spectrum also allows distinguishing between hydrogen and deuterium by optical emission. The wavelength difference between Hα and Dα lines is 0.179 nm (see Figure 7), and this is sufficient for the analysis if a spectrometer with adequate resolution is used [71,72]. In the analysis of H and D in beryllium layers, intensities of only partially resolved Dα and Hα lines measured with the spectral resolution of ~0.1 nm were established by deconvolution, assuming a Gaussian apparatus function and a Lorentzian component caused by Stark broadening [64]. It is worth mentioning that the LIBS signal of hydrogen is enhanced if the analysis is done in helium, typically at a pressure of several torr [72,73]. The likely mechanism of this effect is Penning ionization of hydrogen by the He I, 2s 3S1, and 2s 1S0 metastables (19.82, 20.62 eV), followed by recombination of the H+ ions in the plasma, thereby populating the excited atomic levels from which the analytical hydrogen lines originate [73].
3.1.3. Secondary Ion Mass Spectrometry (SIMS)
Secondary ion mass spectrometry (SIMS) [74,75] provides elemental depth profiles, elemental mapping, and isotopic analysis. The sample surface is sputtered in vacuum with a beam of primary ions (usually O2+ or Cs+), at an energy in the keV range; a small fraction of the sputtered sample material is ionized, and these secondary ions are extracted and analyzed using a mass spectrometer: quadrupole, magnetic sector, or time-of-flight (TOF). Compared to GDOES, SIMS is a much more sensitive technique; SIMS depth profiles may span over several orders of magnitude of the concentrations of some elements. Furthermore, unlike GDOES, elemental mapping is possible, either by direct imaging using ion optics in a magnetic sector instrument or by scanning a finely focused primary beam. Experimentally, SIMS is a more demanding technique than GDOES, as it requires ultra-high-vacuum conditions, especially when analyzing hydrogen, and it is also much more expensive. Unlike GDOES, SIMS is not used for a general bulk analysis of medium and high concentrations of different elements, because of poor accuracy. Typical sputtering rates in SIMS analyses are much lower than in GDOES, typically by a factor of 102, which also affects the duration of analysis. Depth profiling by SIMS is typically by two orders of magnitude slower than GDOES, for the same layer thickness. The same applies to typical analyzed depths. Unlike GDOES, SIMS can be operated in a “static” mode, providing information about topmost atomic layers on the surface. On the other hand, with GDOES, it is easy to analyze layers as thick as ~100 μm, whilst typical depths easily accessible by SIMS are in the range of few hundreds of nanometers only.
The methodology of hydrogen analysis by SIMS was described, e.g., in reviews [76,77]. Cs+ sputtering and detection by H− secondary ions is recommended, while a detection limit of H in silicon of ~2 × 1018 atoms/cm3 was reported in [77], which corresponds to ~40 ppm (atomic) or ~1.4 ppm (weight). It is interesting that this performance was achieved 40 years ago [76] and has not substantially improved ever since. It should also be noted that there is a tradeoff among sensitivity, depth resolution, and lateral resolution [75], and the detection limit mentioned above can hardly be achieved in compositional mapping. SIMS measurements only provide qualitative information, as relative secondary ion intensities are different for different elements and depend on the matrix analyzed and instrumental factors. For quantification, therefore, so-called relative sensitivity factors (RSFs) are required to convert the measured secondary ion intensities into atomic concentrations in the investigated matrix. The RSF is defined as
where i indicates an impurity isotope, M indicates the matrix isotope chosen for reference, ni is the impurity atom density (atoms/cm3) in the matrix, I is the secondary ion intensity (counts/s), and A represents the corresponding isotope abundance. A great effort was devoted in the past to describing the physics of the secondary ion emission. Various physical models were proposed, such as bond-breaking, electron tunneling, and local thermal equilibrium (LTE) models [78]. However, they have not become the basis for a useful quantification scheme for complex matrices or a calibration-free analysis of different elements in the same matrix, a would-be analogy to CF-LIBS. The RSFs of different elements in silicon differ by several orders of magnitude [79]. Only matrix-matched calibrations with dedicated reference samples provide an acceptable analytical performance. In some well-defined matrices of technical importance, such as pure Si or ZnO, RSFs are established using ion implanted standards with known fluences [77]. An example of this approach is the depth profile analysis of hydrogenated amorphous silicon, a-Si:H, using implanted deuterium as internal standard [80]. Concerning the depth scale, virtually no attempts have been reported in the literature to correct for the variable sputtering rate of different layers. Hence, it is very likely that the depth accuracy in the analysis of multilayer systems by SIMS is worse than in GDOES.
Analytical applications involving the analysis of hydrogen by SIMS are diverse, and it is not the intention here to give a comprehensive listing. In the area of semiconductors, in addition to the already mentioned a-Si:H [80], SIMS has been used in several studies to monitor the distribution and migration of hydrogen in heterojunction solar cells, where an intrinsic hydrogen-rich amorphous layer is used to passivate interfacial defects. On annealing, the initially amorphous film of a-SiCx(p):H crystallizes into nanocrystalline nc-SiCx(p), and hydrogen effuses from that layer. Re-hydrogenation can be performed via hydrogen diffusion from a sacrificial layer of SiNx:H [81]. This is an example of a sequence of several technological steps in which hydrogen plays a major role.
In the area of metals, an example of a sophisticated analysis involving 2D SIMS mapping is the study of the redistribution of deuterium in the AISI 304L austenitic steel subjected to external mechanical load [82]. Another example is the analysis of hydrogen diffusion through the oxide layer formed on nickel base alloys in water at a high temperature and high pressure, employing a deuterium tracer [83]. In this study, the SIMS analyses were verified by comparison with elastic recoil detection analysis (ERDA). Depth profiles of 1H and 2D implanted into niobium oxide were reported in [84] (see Figure 8). Analyses by SIMS of hydrogen in materials for nuclear applications involve, e.g., the diffusion of deuterium through a tungsten coating into ferritic steel [85] or through oxide layers formed on Zircalloy-4 and other zirconium alloys [86].
Another application area worth mentioning is the analysis of hydrogen in minerals and rocks. Hydrogen-bearing species of interest in such materials are H2O and OH−. Their analysis can be made using either bulk analysis methods, typically IGF or TDS with IR or mass spectrometric detection (see Section 2), or SIMS, especially if microanalysis or isotope analysis is requested, eventually in combination with FTIR (Fourier-transform infrared spectroscopy). An example is the analysis of volcanic glasses [87,88]. A high-resolution magnetic sector instrument was needed to resolve 18OH ions from 19F. In [87], matrix-specific SIMS calibrations of hydrogen, based on the 1H−/30Si− ratios, were shown for basalt, andesite, and rhyolite. Similar calibrations for olivine, pyroxene, and garnet were presented in [88,89]. A review about the applications of SIMS in geochemistry was provided in [90]. Hydrogen analysis in quartz and quartz glasses was described in [91].
3.2. Nuclear Methods
The methods described in this section employ high-energy ion beams, in the MeV region, and they require instruments and facilities common in nuclear physics, such as particle accelerators, high-energy ion beam optics, and the corresponding detection systems. Such experimental setups are expensive and appear to make these techniques relatively less commonly used for material characterization, but they are nevertheless commercially available. They are frequently used to verify, complement, and help quantify analyses made by other, more easily accessible methods (see, e.g., [37,52,61,83,84,92]).
3.2.1. Elastic Recoil Detection Analysis (ERDA)
In elastic recoil detection analysis (ERDA) [93,94], the sample is irradiated at a glancing angle by MeV ions and the energy of the recoiling target atoms is measured, in the incident hemisphere (forward recoil). Target atoms that are lighter than the incident ions recoil with higher energy than that of forward scattered projectile ions and are, therefore, easier to detect. ERDA is, thus, a suitable method for determination and depth profiling of light isotopes in the presence of heavier elements. This is complementary to a related technique called the Rutherford backscattering spectroscopy (RBS) [93], in which it is the energy spectrum of the scattered projectile ions that carries the analytical information. RBS is suitable for analysis of heavier isotopes in a matrix consisting of lighter atoms. The opposite situation would lead to a high background signal and the consequential deterioration of sensitivity (see Figure 9). In ERDA, ion beams from He to Au with energies in the range 0.3–2 MeV/amu are typically used. The yield of recoiled hydrogen atoms is proportional to the concentration of H and can be established on the basis of the intensity of the incident beam and the geometry of the experiment, using the Rutherford recoil cross-section for H [93]. This makes ERDA a quantitative technique, with a typical accuracy of ~10% relative. If the recoil originates from the surface, the energy Er of the recoil atom is determined only by kinematics (conservation of mass and energy) and can be expressed as
where Ep is the energy of the incident ions, and
where mp and mr represent the mass of the projectile and the recoil atom, respectively, and φ is the angle of detection with respect to the incident beam.
Heavier atoms than H have higher energy Er as per Equation (7) and appear as separate groups in the energy spectrum. If the recoil originates deeper in the sample, the detected energy of the recoil atoms is lower due to the energy loss of the incoming ion and the recoil atom on their paths inside the sample. Therefore, the energy spectrum of recoil atoms also contains information about their depth distribution, which can be used for depth profiling. Systems with an additional time-of-flight detector (TOF-ERDA) have been introduced to support some depth profiling applications [95]. A typical detection limit of ERDA for hydrogen is about 0.1 atomic percent [94]. An example of an ERDA spectrum of an amorphous carbon film with surface contamination by H and O on silicon substrate is shown in Figure 9b.
Hydrogen in carbon films is a frequent application of ERDA. This involves amorphous carbon [96,97] (see Figure 9) and diamond-like films [52,98,99]. H-implanted a-Si [95] and a-Si, a-Ge [100] are examples where hydrogen analysis by ERDA was used in semiconductors. Analysis of hydrogen and deuterium by ERDA in oxide layers on nickel alloys was described in [83]. Analyses of hydrogen in metallic layers by ERDA have also involved, e.g., Al/Cu/Ag/Au multilayers [101], Mg/Ni films [102,103], and Fe/Ti films [104].
3.2.2. Nuclear Reaction Analysis (NRA)
In nuclear reaction analysis (NRA) [105], the sample is bombarded by a beam of accelerated and energy-monochromatized ions that may induce a nuclear reaction with the nuclide in the sample to be analyzed, while one or several particles are emitted, with characteristic energies, and can be used for detection. The most common reaction for hydrogen analysis is the 1H(15N, αγ)12C reaction; the sample is bombarded with 15N ions, which, upon collisions with protons (1H) in the sample, change into carbon 12C, while α and γ particles are emitted, whereby the latter are used for detection. The reaction yield, YB, determines the sensitivity to detect an areal density of hydrogen nuclei (nH (cm−2)) in the sample. It is proportional to the flux (Ni (cm−2·s−1)) of incident projectiles and to the energy-dependent cross-section of the nuclear reaction, σ(Ε) (cm2).
where subscript H denotes hydrogen (1H) and i denotes the incident ion, i.e., 15N. This reaction is resonant; its cross-section σ(Ε) has a narrow peak at ER = 6.385 MeV (the next resonance of the 1H(15N, αγ)12C reaction occurs at 13.35 eV), with a width of only 1.8 keV. The ratio of the on-resonance to the nearby off-resonance cross-section is >104. This makes this reaction very suitable for depth profiling; if the incident ions have a higher energy than the resonance energy, they are decelerated on their path through the sample material and reach the resonance energy at a certain depth. The reaction then proceeds only in a very narrow layer around that particular depth, see Figure 4b. Typical stopping powers S = (dE/dz) for 15N near the 6.4 MeV range are on the order of ~1–4 keV/nm for elemental targets; thus, the typical near-surface depth resolution is between ~1 and a few nanometers. At larger depths, depth resolution worsens, which is a phenomenon referred to as straggling [105]. The stopping power for a given matrix can be calculated on the basis of the areal number densities (cm−2) of the elements present in the matrix and their stopping cross-sections ε (for 15N at 6.385 MeV). Those are known and tabulated, e.g., in [105]. The stopping power S is required not only to convert the energy abscissa of the NRA spectrum into the probing depth but also to derive absolute H concentrations from γ-yield measurements at bulk targets. An extensive collection of experimental (dE/dz) values is, e.g., the database in [106]. For analytical interpretation of NRA measurements, dedicated reference materials have also been proposed, e.g., hydrogenated tantalum [107]. The 1H(15N, αγ)12C reaction provides good sensitivity, with detection limits around ~100 atomic ppm H or even lower, as well as a practical probing range (2–4 μm). There are few other nuclear reactions for the analysis of 1H [105]; however, the 1H(15N, αγ)12C reaction provides generally the best performance and is most widely used. It should also be noted that two NRA systems for depth-resolved two-dimensional hydrogen mapping have been developed in Japan [108], providing lateral resolution of 30 and 150 μm.
A comparison of NRA (the 1H(15N, αγ)12C nuclear reaction) and ERDA was presented in [105]. NRA seems to outperform ERDA in terms of depth resolution and sensitivity (on the order of 10 weight ppm for NRA vs. 50–100 for ERDA). Furthermore, the processing of NRA data requires the use of less complex models (particle–matter interaction) than ERDA to extract distribution maps or H concentration profiles. However, in terms of lateral resolution, because of the size of the primary beam, ERDA outperforms the micro-beam NRA.
In addition to the 1H isotope, a common NRA technique exists for deuterium, namely, the D(3He, p)4He reaction [109]. It produces protons of ~13 MeV and α particles with 2–4 MeV energy that are easily identified. The D(3He, p)4He cross-section has a broad (~350 keV) peak at 650 keV. Since the stopping powers for the product α particles and protons are small, the reaction offers a very poor depth resolution (~500 nm near the surface). The large resonance width allows quantifying the D content in the entire near-surface region [109].
NRA is a mature method and has been used in many application areas. It is often combined with ERDA to get more complete information. Hydrogen analysis by NRA in metals has involved, e.g., Al/Cu/Ag/Au multilayers [101], Mg/Ni films [102], tantalum [107], Zr/Cu/Al glassy alloys [108], and deuterium in W and WO3 layers [61], in niobium [84,92], and beryllium [110]. Concerning semiconductors, NRA has been used, e.g., to analyze hydrogen in a-Si:H [37,111], silicon carbide [112], and silicon nitride [113]. Nonmetals are represented, e.g., by the analysis of deuterium implanted into carbon [114] and hydrogen analysis in diamond-like carbon coatings [115]. A number of other applications of NRA to hydrogen analysis were listed in [105].
4. Conclusions and Perspectives
Hydrogen is an element that affects many areas of science and technology, ranging from unwanted effects in classical metallurgy to electrochemical systems, corrosion of metals, the development of hydrogen-storage materials, semiconductors, various coatings, and thin-film systems. Hydrogen is also of interest in geochemistry, nuclear technologies, and many other areas. This yields an ever-increasing demand for the analysis of hydrogen in a very diverse range of materials, processes, and applications. The currently used bulk analysis and depth profiling methods for hydrogen in inorganic materials and inorganic coatings were reviewed in this paper. The bulk analysis methods of hydrogen are based on fusion or controlled heating of macroscopic samples and the detection of hydrogen, H2, in the released gases. They offer excellent accuracy and sensitivity. Depth profiling methods involve optical emission spectroscopy (GDOES and LIBS), mass spectrometry (GDMS and SIMS), and nuclear methods (ERDA and NRA). The principles of all these methods were explained in terms of their methodology, calibration procedures, analytical performance, and major application areas. The synergies and the complementarity of various methods of hydrogen analysis were described. The existing literature about all these methods was categorized and critically evaluated, while major papers concerning each method were selected and listed, in the context relevant to every aspect mentioned in the review.
Lastly, a few general remarks about hydrogen analysis should be made. Virtually all the methods listed here have been around for decades. Some have been developed to such a level that they can be considered as mature, with only little potential for further major improvements. This applies, e.g., to bulk analysis methods (Section 2). Concerning emission spectroscopies (Section 3.1.1 and Section 3.1.2), a major issue is not the experiment itself but rather the interpretation of the data. In GDOES and GDMS, a number of processes involving hydrogen occur in the glow discharge plasma that are far from being completely described and understood. However, they significantly affect the measured intensities [43,44,45,46,47]. Likewise, in LIBS, analytical interpretation of the spectra is a very complex task, especially in the calibration-free approach. Further work on the fundamentals of these methods is desirable. In nuclear methods, significant instrumental developments have occurred relatively recently, such as hydrogen mapping by NRA.
Interesting is also the point of view of applications. Whereas the bulk analysis of hydrogen is considered rather routine and the relevant instrument manufacturers (e.g., LECO, Bruker, Eltra, Horiba, and some others) keep extensive libraries of application notes for a wide range of materials, for the methods listed in Section 3, the methodology for a particular analytical task is usually nontrivial and is, therefore, more frequently described in the scientific literature. In the papers focusing primarily on the coating under study, as well as its preparation, properties, etc., analytical aspects are sometimes treated incompletely, the results are often interpreted without considering the uncertainty of the analyses, etc. More attention to such aspects is usually paid in journals devoted to analytical science or instrumentation. A very fruitful approach to the analysis of coatings, particularly of hydrogen, is when the analytical methodology is developed hand in hand with the process itself, e.g., by preparing hydrogen-containing sample(s) that can be characterized by an independent method and subsequently used as a reference in more routine analyses, when optimizing the deposition technology itself.
Funding
This work was supported by the Operational Program “Research, Development, and Education” financed by European Structural and Investment Funds and the Czech Ministry of Education, Youth, and Sports (Project No. SOLID21-CZ.02.1.01/0.0/0.0/16_019/0000760).
Conflicts of Interest
The author declares no conflict of interest.
References
- Alefeld, G.; Völkl, J. (Eds.) Hydrogen in Metals II: Application-Oriented Properties; Springer: Berlin/Heidelberg, Germany, 1978. [Google Scholar]
- Fukai, Y. The Metal-Hydrogen System; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Smialowski, M. Hydrogen in Steel: Effect of Hydrogen on Iron and Steel during Production, Fabrication and Use; Pergamon: Oxford, UK, 1962. [Google Scholar]
- Hydrogen Embrittlement. Available online: https://en.wikipedia.org/wiki/Hydrogen_embrittlement (accessed on 3 February 2021).
- Świerczyńska, A.; Fydrych, D.; Landowski, M.; Rogalski, G.; Łabanowski, J. Hydrogen embrittlement of X2CrNiMoCuN25-6-3 super duplex stainless steel welded joints under cathodic protection. Constr. Build. Mater. 2020, 238, 117697. [Google Scholar] [CrossRef]
- Fan, J.; Chen, H.; Zhao, W.; Yan, L. Study on flake formation behavior and its influence factors in Cr5 steel. Materials 2018, 11, 690. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Su, Y.J.; Chu, W.Y.; Li, Z.B.; Liang, D.T.; Qiao, L.J. Hydrogen embrittlement of weld metal of austenitic stainless steels. Corros. Sci. 2002, 44, 1983–1993. [Google Scholar] [CrossRef]
- Padhy, G.K.; Komizo, Y.I. Diffusible hydrogen in steel weldments: A status review. Trans. JWRI 2013, 42, 39–62. [Google Scholar]
- Fydrych, D.; Łabanowski, J. Determining diffusible hydrogen amounts using the mercury method. Weld. Int. 2012, 26, 697–702. [Google Scholar] [CrossRef]
- Enomoto, M.; Cheng, L.; Mizuno, H.; Watanabe, Y.; Omura, T.; Sakai, J.I.; Yokoyama, K.I.; Suzuki, H.; Okuma, R. Hydrogen Absorption into Austenitic Stainless Steels Under High-Pressure Gaseous Hydrogen and Cathodic Charge in Aqueous Solution. Metall. Mater. Trans. E 2014, 1, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Fournier, L.; Delafosse, D.; Magnin, T. Cathodic hydrogen embrittlement in alloy 718. Mater. Sci. Eng. 1999, 269, 111–119. [Google Scholar] [CrossRef]
- Ambat, R.; Dwarakadasa, E.S. Effect of hydrogen in aluminium and aluminium alloys: A review. Bull. Mater. Sci. 1996, 19, 103–114. [Google Scholar] [CrossRef]
- Anyalebechi, P.N. Techniques for determination of the Hydrogen Content in Aluminium and its Alloys—A Review. Cast Met. 1990, 3, 182–201. [Google Scholar] [CrossRef]
- Froes, F.H.; Eliezer, D.; Moody, N.R. Hydrogen Effects in Titanium, a Chapter in Hydrogen Effects in Materials; Thompson, A.W., Moody, N.R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Kamilyan, M.; Silverstein, R.; Eliezer, D. Hydrogen trapping and hydrogen embrittlement of Mg alloys. J. Mater. Sci. 2017, 52, 11091–11100. [Google Scholar] [CrossRef]
- Talbot, D.E.J. Effects of Hydrogen in Aluminium, Magnesium, Copper, and Their Alloys. Int. Metall. Rev. 1975, 20, 166–184. [Google Scholar] [CrossRef]
- Dekura, S.; Kobayashi, H.; Kusada, K.; Kitagawa, H. Hydrogen in Palladium and Storage Properties of Related Nanomaterials: Size, Shape, Alloying, and Metal-Organic Framework Coating Effects. ChemPhysChem 2019, 20, 1158–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alloyed Steel No. 1.4546.9 with Certified Hydrogen Content. Available online: https://rrr.bam.de/RRR/Navigation/EN/Reference-Materials/reference-materials.html (accessed on 11 March 2021).
- Xie, Z.K.; Ikeda, T.; Okuda, Y.; Nakajima, H. Characteristics of Sound Absorption in Lotus-Type Porous Magnesium. Jpn. J. Appl. Phys. 2004, 43, 7315–7319. [Google Scholar] [CrossRef]
- Lawrenz, D.; Mitchell, J. Thermal Evolution Methods for Carbon, Sulfur, Oxygen, Nitrogen and Hydrogen in Iron and Steel Analysis. In Encyclopedia of Analytical Chemistry, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp. 8991–9008. [Google Scholar]
- Thermal Conductivity Detector. Available online: https://en.wikipedia.org/wiki/Thermal_conductivity_detector (accessed on 13 March 2021).
- Littlewood, A.B. Sensitivity of Catharometers in Gas Chromatography and the Thermal Conductivity of Binary Gas Mixtures. Nature 1959, 184, 1631–1632. [Google Scholar] [CrossRef]
- Dinh, T.-V.; Choi, I.-Y.; Son, Y.-S.; Kim, J.-C. A review on non-dispersive infrared gas sensors: Improvement of sensor detection limit and interference correction. Sens. Actuators B Chem. 2016, 231, 529–538. [Google Scholar] [CrossRef]
- Silverstein, R.; Eliezer, D.; Tal-Gutelmacher, E. Hydrogen trapping in alloys studied by thermal desorption spectrometry. J. Alloys Compd. 2018, 747, 511–522. [Google Scholar] [CrossRef]
- Tal-Gutelmacher, E.; Eliezer, D.; Abramov, E. Thermal desorption spectroscopy (TDS)—Application in quantitative study of hydrogen evolution and trapping in crystalline and non-crystalline materials. Mater. Sci. Eng. A 2007, 445–446, 625–631. [Google Scholar] [CrossRef]
- Rhode, M.; Schaupp, T.; Muenster, C.; Mente, T.; Boellinghaus, T.; Kannengiesser, T. Hydrogen determination in welded specimens by carrier gas hot extraction—A review on the main parameters and their effects on hydrogen measurement. Weld. World 2019, 63, 511–526. [Google Scholar] [CrossRef]
- Bergers, K.; de Souza, E.C.; Thomas, I.; Mabho, N.; Flock, J. Determination of Hydrogen in Steel by Thermal Desorption Mass Spectrometry. Steel Res. Int. 2010, 81, 499–507. [Google Scholar] [CrossRef]
- Georges, C.; Sturel, T.; Drillet, P.; Mataigne, J.-M. Absorption/Desorption of Diffusible Hydrogen in Aluminized Boron Steel. ISIJ Int. 2013, 53, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, M.; Mitzschke, N.; Jüttner, S. Determination of Hydrogen Transport Behaviour in Boron-Manganese Steels Using Different Methods and Boundary Conditions. Metals 2019, 9, 1007. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lin, H.J.; Cai, W.T.; Ouyang, L.Z.; Zhu, M. Tuning kinetics and thermodynamics of hydrogen storage in light metal element-based systems—A review of recent progress. J. Alloys Compd. 2016, 658, 280–300. [Google Scholar] [CrossRef]
- Babikhina, M.N.; Kudiiarov, V.N.; Mostovshchikov, A.V.; Lider, A.M. Quantitative and Qualitative Analysis of Hydrogen Accumulation in Hydrogen-Storage Materials Using Hydrogen Extraction in an Inert Atmosphere. Metals 2018, 8, 672. [Google Scholar] [CrossRef] [Green Version]
- Titanium Hydride, Wikipedia. Available online: https://en.wikipedia.org/wiki/Titanium_hydride (accessed on 20 March 2021).
- Lisowski, W.; Keim, E.G.; Kaszkur, Z.; Smithers, M.A. Decomposition of thin titanium deuteride films; thermal desorption kinetics studies combined with microstructure analysis. Appl. Surf. Sci. 2008, 254, 2629–2637. [Google Scholar] [CrossRef]
- Ashcheulov, P.; Taylor, A.; Vlčková-Živcová, Z.; Hubík, P.; Honolka, J.; Vondráček, M.; Remzová, M.; Kopeček, J.; Klimša, L.; Lorinčik, J.; et al. Low temperature synthesis of transparent conductive boron doped diamond films for optoelectronic applications: Role of hydrogen on the electrical properties. Appl. Mater. Today 2020, 19, 10063. [Google Scholar]
- Marcus, R.K.; Broekaert, J.A.C. (Eds.) Glow Discharge Plasmas in Analytical Spectroscopy; John Wiley & Sons: New York, NY, USA, 2003. [Google Scholar]
- Lobo, L.; Fernandez, B.; Pereiro, R. Depth profile analysis with glow discharge spectrometry. J. Anal. At. Spectrom. 2017, 32, 920–930. [Google Scholar] [CrossRef]
- Steffens, J.; Becker, H.-W.; Gerke, S.; Joos, S.; Hahn, G.; Terheiden, B. Replacing NRA by fast GD-OES measurements as input to a model-based prediction of hydrogen diffusion in a-Si. Energy Procedia 2017, 124, 180–187. [Google Scholar] [CrossRef]
- Alvarez-Toral, A.; Sanchez, P.; Menéndez, A.; Pereiro, R.; Sanz-Medel, A.; Fernández, B. Depth Profile Analysis of Amorphous Silicon Thin Film Solar Cells by Pulsed Radiofrequency Glow Discharge Time of Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 305–314. [Google Scholar] [CrossRef]
- Weiss, Z. Calibration methods in glow discharge optical emission spectroscopy: A tutorial review. J. Anal. At. Spectrom. 2015, 30, 1038–1049. [Google Scholar] [CrossRef]
- Gonzalez-Gago, C.; Bordel, N.; Pisonero, J. Glow Discharge Mass Spectrometry. In ASM Handbook, Materials Characterization; ASM International: Novelty, OH, USA, 2019; Volume 10, pp. 153–161. [Google Scholar]
- Gonzalez-Gago, C.; Šmíd, P.; Hofmann, T.; Venzago, C.; Hoffmann, V.; Gruner, W.; Pfeifer, J.; Richter, S.; Kipphardt, H. Investigations of matrix independent calibration approaches in fast flow glow discharge mass spectrometry. J. Anal. At. Spectrom. 2019, 34, 1109–1124. [Google Scholar] [CrossRef]
- Schubert, C.; Hoffmann, V.; Kümmel, A.; Sinn, J.; Härtel, M.; Reuther, A.; Thomalla, M.; Gemming, T.; Eckert, J.; Leyens, C. Compositional depth profiling of diamond-like carbon layers by glow discharge optical emission spectroscopy. J. Anal. At. Spectrom. 2016, 31, 2207–2212. [Google Scholar] [CrossRef] [Green Version]
- Bengtson, A.; Hänström, S. The influence of hydrogen on emission intensities in GD-OES, consequences for quantitative depth profile analysis. In Proceedings of the 5th International Conference on Progress in Analytical Chemistry in the Steel and Metals (CETAS), Luxembourg, 12–14 May 1998; pp. 47–54. [Google Scholar]
- Šmíd, P.; Steers, E.B.M.; Weiss, Z.; Pickering, J.C.; Hoffmann, V. The effect of hydrogen and nitrogen on emission spectra of iron and titanium atomic lines in analytical glow discharges. J. Anal. At. Spectrom. 2008, 23, 1223–1233. [Google Scholar] [CrossRef]
- Steers, E.B.M.; Smid, P.; Weiss, Z. Asymmetric charge transfer with hydrogen ions—An important factor in the “hydrogen effect” in glow discharge optical emission spectroscopy. Spectrochim. Acta Part B 2006, 61, 414–420. [Google Scholar] [CrossRef]
- Mushtaq, S.; Steers, E.B.M.; Whitby, J.A.; Horvath, P.; Michler, J.; Pickering, J.C. A glow discharge time-of flight mass spectrometry (GD-TOFMS) study of the ‘hydrogen effect’ using copper, iron and titanium cathodes. J. Anal. At. Spectrom. 2015, 30, 1774–1781. [Google Scholar] [CrossRef]
- Weiss, Z.; Vlcak, P. Analysis of shallow depth profiles of titanium nitride and N-implanted titanium by GD-OES: The ‘hydrogen effect’ after the discharge startup and a correction thereof. J. Anal. At. Spectrom. 2017, 32, 2476–2484. [Google Scholar] [CrossRef]
- Hoffmann, V.; Uhlemann, M.; Richter, S.; Pfeifer, J. Calibration capacity of hot-pressed hydrogen standards for glow discharge optical emission and mass spectrometry. Spectrochim. Acta Part B 2021, 176, 106039. [Google Scholar] [CrossRef]
- Priamusho, T.S.; Mikhaylov, A.A.; Babikhina, M.N.; Kudiiarov, V.N.; Laptev, R.S. Glow Discharge Optical Emission Spectrometer Calibration Using Hydrogenated Zr-2.5Nb Alloy Standard Samples. Metals 2018, 8, 372. [Google Scholar] [CrossRef] [Green Version]
- Hodoroaba, V.-D.; Klemm, D.; Reinholz, U.; Strub, E.; Röhrich, J.; Bohne, W.; Hoffmann, V.; Wetzig, K. Potential candidates of certified reference material for determination of hydrogen concentration with glow discharge optical emission spectrometry (GD-OES)—A feasibility study. J. Anal. At. Spectrom. 2008, 23, 460–462. [Google Scholar] [CrossRef]
- Wilke, M.; Teichert, G.; Gemma, R.; Pundt, A.; Kirchheim, R.; Romanus, H.; Schaaf, P. Glow discharge optical emission spectroscopy for accurate and well resolved analysis of coatings and thin films. Thin Solid Film 2011, 520, 1660–1667. [Google Scholar] [CrossRef]
- Takahara, H.; Ishigami, R.; Kodama, K.; Kojyo, A.; Nakamura, T.; Oka, Y. Hydrogen analysis in diamond-like carbon by glow discharge optical emission spectroscopy. J. Anal. At. Spectrom. 2016, 31, 940–947. [Google Scholar] [CrossRef]
- Richter, K.; Waldmann, T.; Kasper, M.; Pfeifer, C.; Memm, M.; Axmann, P.; Wohlfahrt-Mehrens, M. Surface Film Formation and Dissolution in Si/C Anodes of Li-Ion Batteries: A Glow Discharge Optical Emission Spectroscopy Depth Profiling Study. J. Phys. Chem. C 2019, 123, 18795–18803. [Google Scholar] [CrossRef]
- Lang, E.; Taylor, C.N.; Allain, J.P. GD-OES study of the influence of second phase particles on the deuterium depth distribution in dispersion-strengthened tungsten. J. Nucl. Mater. 2020, 532, 152047. [Google Scholar] [CrossRef]
- Qiao, L.; Zhang, X.; He, R.; Zhang, H.; Fu, E.; Wang, P. Experimental measurement of deuterium concentration and depth profiling in tungsten by radio frequency glow discharge optical emission spectroscopy. Spectrochim. Acta Part B 2020, 173, 105975. [Google Scholar] [CrossRef]
- Efimov, A.; Kasik, M.; Putyera, K.; Moreau, O. Measurement of Hydrogen and Deuterium Concentration in Gold Electroplated Layer by Glow Discharge Mass Spectrometry. Electrochem. Solid State Lett. 2000, 3, 477–478. [Google Scholar] [CrossRef]
- Cremers, D.A.; Radziemski, L.J. Handbook of Laser-Induced Breakdown Spectroscopy, 2nd ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Joliveta, L.; Leprince, M.; Moncayo, S.; Sorbier, L.; Lienemann, C.-P.; Motto-Ros, V. Review of the recent advances and applications of LIBS-based imaging. Spectrochim. Acta Part B 2019, 151, 41–53. [Google Scholar] [CrossRef]
- Tognoni, E.; Cristoforetti, G.; Legnaioli, S.; Palleschi, V. Calibration-free laser-induced breakdown spectroscopy: State of the art. Spectrochim. Acta Part B 2010, 65, 1–14. [Google Scholar] [CrossRef]
- Roldán, A.M.; Pisarčík, M.; Veis, M.; Držík, M.; Veis, P. Calibration-free analysis of a tungsten-based target for diagnostics of relevant fusion materials comparing picosecond and nanosecond LIBS. Spectrochim. Acta Part B 2021, 177, 106055. [Google Scholar] [CrossRef]
- Paris, P.; Jõgi, I.; Piip, K.; Passoni, M.; Dellasega, D.; Grigore, E.; Arnoldbik, W.M.; van der Meiden, H. In-situ LIBS and NRA deuterium retention study in porous W-O and compact W coatings loaded by Magnum-PSI. Fusion Eng. Des. 2021, 168, 112403. [Google Scholar] [CrossRef]
- Aragón, C.; Aguilera, J.A. Quantitative analysis by laser-induced breakdown spectroscopy based on generalized curves of growth. Spectrochim. Acta Part B 2015, 110, 124–133. [Google Scholar] [CrossRef]
- Ashikawa, N.; Zhao, D.; Li, C.; Ding, H.; LHD Experimental Group. Hydrogen Depth Profiles Using Laser-Induced Breakdown Spectroscopy (LIBS) on Graphite Target of Divertor in LHD, Proc. of A3 Foresight Program Seminar on Critical Physics Issues Specific to Steady State Sustainment of High-Performance Plasmas 2015. Available online: https://inis.iaea.org/collection/NCLCollectionStore/_Public/49/089/49089674.pdf?r=1 (accessed on 28 March 2021).
- Dwivedi, V.; Marín-Roldán, A.; Karhunen, J.; Paris, P.; Jõgi, I.; Porosnicu, C.; Lungu, C.P.; van der Meiden, H.; Hakola, A.; Veis, P. CF-LIBS quantification and depth profile analysis of Be coating mixed layers. Nucl. Mater. Energy 2021, 27, 100990. [Google Scholar] [CrossRef]
- Kautz, E.J.; Devaraj, A.; Senor, D.J.; Harilal, S.S. Hydrogen isotopic analysis of nuclear reactor materials using ultrafast laser-induced breakdown spectroscopy. Opt. Express 2001, 29, 4936–4946. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.; Landis, G.P.; Maroef, I.; Olson, D.L.; Wildeman, T.R. The determination of hydrogen distribution in high-strength steel weldments Part 1: Laser ablation methods. Weld. J. 2001, 80, 115s–121s. [Google Scholar]
- Imashuku, S.; Kamimura, T.; Kashiwakura, S.; Wagatsuma, K. Quantitative Analysis of Hydrogen in High-Hydrogen-Content Material of Magnesium Hydride via Laser-Induced Breakdown Spectroscopy. Anal. Chem. 2020, 92, 11171–11176. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Yang, R.; Li, Q.; Ye, X.; Wu, J.; Chen, C.; Wang, X.; Chen, X. Quantitative measurement of hydrogen isotopes in titanium using laser-induced breakdown spectroscopy. Appl. Opt. 2020, 59, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, Q.; Yang, R.; Cui, X. Quantitative determination of hydrogen isotope in titanium using LIBS. In AOPC 2019: Optical Spectroscopy and Imaging, Proceedings of the Applied Optics and Photonics China (AOPC2019), Beijing, China, 7–9 July 2019; Proc. SPIE—International Society for Optics and Photonics: Bellingham, WA, USA, 2019; Volume 11337, p. 12. [Google Scholar] [CrossRef]
- Thomas, N.H.; Ehlmann, B.L.; Anderson, D.E.; Clegg, S.M.; Forni, O.; Schröder, S.; Rapin, W.; Meslin, P.-Y.; Lasue, J.; Delapp, D.M.; et al. Characterization of Hydrogen in Basaltic Materials With Laser-Induced Breakdown Spectroscopy (LIBS) for Application to MSL ChemCam Data. J. Geophys. Res. Planets 2018, 123, 1996–2021. [Google Scholar] [CrossRef] [Green Version]
- Kurniawan, K.H.; Tjia, M.O.; Kagawa, K. Review of Laser-Induced Plasma, Its Mechanism, and Application to Quantitative Analysis of Hydrogen and Deuterium. Appl. Spectr. Rev. 2014, 49, 323–434. [Google Scholar] [CrossRef]
- Kurniawan, K.H.; Lie, T.J.; Suliyanti, M.M.; Hedwig, R.; Pardede, M.; Kurniawan, D.P.; Kusumoto, Y.; Kagawa, K. Quantitative analysis of deuterium using laser-induced plasma at low pressure of helium. Anal. Chem. 2006, 78, 5768–5773. [Google Scholar] [CrossRef]
- Pardede, M.; Lie, T.J.; Iqbal, J.; Bilal, M.; Hedwig, R.; Ramli, M.; Khumaeni, A.; Budi, W.S.; Idris, N.; Abdulmadjid, S.N. H–D analysis employing energy transfer from metastable excited-state He in double-pulse LIBS with low-pressure He gas. Anal. Chem. 2019, 91, 1571–1577. [Google Scholar] [CrossRef]
- Walker, A.V. Secondary Ion Mass Spectrometry, chapter in Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 44–49. [Google Scholar]
- Van der Heide, P. Secondary Ion Mass Spectrometry: An Introduction to Principles and Practices; John Wiley and Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Magee, C.W.; Botnick, E.M. Hydrogen depth profiling using SIMS—Problems and their solutions. J. Vac. Sci. Technol. 1981, 19, 47–52. [Google Scholar] [CrossRef]
- Zhu, Z.; Shutthanandan, V.; Engelhard, M. An investigation of hydrogen depth profiling using ToF-SIMS. Surf. Interface Anal. 2012, 44, 232–237. [Google Scholar] [CrossRef]
- Chakraborty, P. Secondary ion mass spectrometry for quantitative surface and in-depth analysis of materials. Pramana J. Phys. 1998, 50, 617–640. [Google Scholar] [CrossRef]
- Wilson, R.G.; Stevie, F.A.; Magee, C.W. Secondary Ion Mass Spectrometry: A Practical Handbook for Depth Profiling and Bulk Impurity Analysis; John Wiley & Sons: Hoboken, NJ, USA, 1989. [Google Scholar]
- Williams, P.; Stika, K.M.; Davies, J.A.; Jackman, T.E. Quantitative SIMS analysis of hydrogenated amorphous silicon using superimposed deuterium implant standards. Nucl. Instrum. Methods 1983, 218, 299–302. [Google Scholar] [CrossRef]
- Lehmanna, M.; Valle, N.; Horzel, J.; Pshenova, A.; Wyss, P.; Döbeli, M.; Despeisse, M.; Eswara, S.; Wirtz, T.; Jeangros, Q.; et al. Analysis of hydrogen distribution and migration in fired passivating contacts (FPC). Sol. Energy Mater. Solar Cells 2019, 200, 110018. [Google Scholar] [CrossRef] [Green Version]
- Röhsler, A.; Sobol, O.; Hänninen, H.; Böllinghaus, T. In-situ ToF-SIMS analyses of deuterium re-distribution in austenitic steel AISI 304L under mechanical load. Sci. Rep. 2020, 10, 3611. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Seyeux, A.; Vickridge, I.; Voyshnis, S.; Marcus, P. ToF-SIMS and ERDA study of hydrogen and deuterium in nickel-base alloys oxidized in water. Corros. Sci. 2018, 140, 151–158. [Google Scholar] [CrossRef]
- Stevie, F.A. Analysis of hydrogen in materials with and without high hydrogen mobility. Surf. Interface Anal. 2016, 48, 310–314. [Google Scholar] [CrossRef]
- Xu, Y.; Hirooka, Y.; Luo, L.M.; Wu, Y.C. Deuterium concentration depth profiling in sputter-deposited tungsten coated F82H using secondary ion mass spectrometry. Nucl. Mater. Energy 2019, 21, 100708. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Sayers, J.; Aarholt, T.; He, G.; Hulme, H.; Garner, A.; Preuss, M.; Nordin, H.; Partezana, J.M.; et al. Characterisation of deuterium distributions in corroded zirconium alloys using high-resolution SIMS imaging. Acta Mater. 2020, 200, 581–596. [Google Scholar] [CrossRef]
- Hauri, E.; Wang, J.; Dixon, J.E.; King, P.L.; Mandeville, C.; Newman, S. SIMS analysis of volatiles in silicate glasses 1. Calibration, matrix effects and comparisons with FTIR. Chem. Geol. 2002, 183, 99–114. [Google Scholar] [CrossRef]
- Koga, K.; Hauri, E.H.; Hirschmann, M.M.; Bell, D. Hydrogen concentration analyses using SIMS and FTIR: Comparison and calibration for nominally anhydrous minerals. Geochem. Geophys. Geosyst. 2003, 4, 1019. [Google Scholar] [CrossRef] [Green Version]
- Mosenfelder, J.L.; le Voyer, M.; Rossman, G.R.; Guan, Y.; Bell, D.R.; Asimow, P.D.; Eiler, J.M. Analysis of hydrogen in olivine by SIMS: Evaluation of standards and protocol. Am. Mineral. 2011, 96, 1725–1741. [Google Scholar] [CrossRef]
- Shimizu, N.; Hart, S.R. Application of the ion microprobe to geochemistry and cosmochemistry. Annu. Rev. Earth Planet. Sci. 1982, 10, 483–526. [Google Scholar] [CrossRef]
- Yurimoto, H.; Kurosawa, M.; Sueno, S. Hydrogen analysis in quartz crystals and quartz glasses by secondary ion mass spectrometry. Geochim. Cosmochim. Acta 1989, 53, 751–755. [Google Scholar] [CrossRef]
- Maheshwari, P.; Stevie, F.A.; Myneni, G.; Ciovati, G.; Rigsbee, J.M.; Griffis, D.P. Analysis of Interstitial Elements in Niobium with Secondary Ion Mass Spectrometry (SIMS). In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 2011; Volume 1352, pp. 151–160. [Google Scholar]
- Cohen, D.D.; Bird, R.; Dytlewski, N.; Siegele, R. Ion Beams for Material Analysis. In Encyclopedia of Physical Science and Technology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 55–63. [Google Scholar]
- Jain, I.P.; Jain, A.; Jain, P. ERDA: Technique for Hydrogen Content and Depth Profile in Thin Film Metal Hydride. In Proceedings of the 18th World Hydrogen Energy Conference 2010, Essen, Germany, 16–21 May 2010; Available online: https://www.osti.gov/etdeweb/servlets/purl/21400976 (accessed on 5 April 2021).
- Siketić, Z.; Radović, I.B.; Jakšić, M. Quantitative analysis of hydrogen in thin films using time-of-flight recoil detection analysis. Thin Solid Film 2010, 518, 2617–2622. [Google Scholar] [CrossRef]
- Tunmee, S.; Supruangnet, R.; Nakajima, H.; Zhou, X.; Arakawa, S.; Suzuki, T.; Kanda, K.; Ito, H.; Komatsu, K.; Saitoh, H. Study of Synchrotron Radiation Near-Edge X-Ray Absorption Fine-Structure of Amorphous Hydrogenated Carbon Films at Various Thicknesses. J. Nanomater. 2015, 2015, 276790. [Google Scholar] [CrossRef]
- Gago, R.; Jiménez, I.; Albella, J.M.; Climent-Font, A.; Banks, J.C.; Doyle, B.L.; Terminello, J.L. Bonding and hardness in nonhydrogenated carbon films with moderate sp3 content. J. Appl. Phys. 2000, 87, 8174–8180. [Google Scholar] [CrossRef] [Green Version]
- Ouchabane, M.; Salah, H.; Herrmann, M.; Tabet, N.; Henda, K.; Touchrift, B.; Kechouane, M. Influence of bias voltage on the structure and deposition mechanism of diamond-like carbon films produced by RF (13.56 MHz) CH4 plasma. Phys. Status Solidi A 2010, 207, 2311–2318. [Google Scholar] [CrossRef]
- Čekada, M.; Kahn, M.; Pelicon, P.; Siketić, Z.; Radović, I.B.; Waldhauser, W.; Paskvale, S. Analysis of nitrogen-doped ion-beam-deposited hydrogenated diamond-like carbon films using ERDA/RBS, TOF-ERDA and Raman spectroscopy. Surf. Coat. Technol. 2012, 211, 72–75. [Google Scholar] [CrossRef]
- Khánh, N.Q.; Serényi, M.; Csik, A.; Frigeri, C. Determination of hydrogen concentration in a-Si and a-Ge layers by elastic recoil detection analysis. Vacuum 2012, 86, 711–713. [Google Scholar] [CrossRef] [Green Version]
- Wielunski, L.S.; Grambole, D.; Kreissig, U.; Grötzschel, R.; Harding, G.; Szilágyi, E. Hydrogen depth resolution in multilayer metal structures, comparison of elastic recoil detection and resonant nuclear reaction method. Nucl. Instrum. Methods Phys. Res. B 2002, 190, 693–698. [Google Scholar] [CrossRef]
- Pranevicius, L.; Wirth, E.; Milcius, D.; Lelis, M.; Pranevicius, L.L.; Bacianskas, A. Structure transformations and hydrogen storage properties of co-sputtered MgNi films. Appl. Surf. Sci. 2009, 255, 5971–5974. [Google Scholar] [CrossRef]
- Wirth, E.; Munnik, F.; Pranevicius, L.L.; Milcius, D. Dynamic surface barrier effects on hydrogen storage capacity in Mg–Ni films. J. Alloys Compd. 2009, 475, 917–922. [Google Scholar] [CrossRef]
- Jain, I.P.; Devi, B.; Sharma, P.; Williamson, A.; Vijay, Y.K.; Avasthi, D.K.; Tripathi, A. Hydrogen in FeTi thin films by ERDA with Ag107 ions. Int. J. Hydrog. Energy 2000, 25, 517–521. [Google Scholar] [CrossRef]
- Wilde, M.; Fukutani, K. Hydrogen detection near surfaces and shallow interfaces with resonant nuclear reaction analysis. Surf. Sci. Rep. 2014, 69, 196–295. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, J.F.; Biersack, J.P.; Littmark, U. Stopping and Ranges of Ions in Solids; Pergamon: New York, NY, USA, 1985. [Google Scholar]
- Hjörvarsson, B.; Rydén, J.; Ericsson, T.; Karlsson, E. Hydrogenated tantalum: A convenient calibration substance for hydrogen profile analysis using nuclear resonance reactions. Nucl. Instrum. Methods Phys. Res. B 1989, 42, 257–263. [Google Scholar] [CrossRef]
- Sekiba, D.; Yonemura, H.; Ogura, S.; Matsumoto, M.; Kitaoka, Y.; Yokoyama, Y.; Matsuzaki, H.; Narusawa, T.; Fukutani, K. Development of micro-beam NRA for hydrogen mapping: Observation of fatigue-fractured surface of glassy alloys. Nucl. Instrum. Methods Phys. Res. B 2011, 269, 627–631. [Google Scholar] [CrossRef]
- Dieumegard, D.; Dubreuil, D.; Amsel, G. Analysis and depth profiling of deuterium with the D(3He, p)4He reaction by detecting the protons at backward angles. Nucl. Instrum. Methods 1979, 166, 431–445. [Google Scholar] [CrossRef]
- Wampler, W.R. Trapping of deuterium in beryllium. J. Nucl. Mater. 1992, 196–198, 981–985. [Google Scholar] [CrossRef]
- Danesh, P.; Pantchev, B.; Schmidt, B. Infrared absorption strengths of ion-implanted hydrogenated amorphous silicon. Thin Solid Film 2008, 516, 3383–3386. [Google Scholar] [CrossRef]
- Portmann, J.; Haug, C.; Brenn, R.; Schneider, J.; Rottner, K.; Helbig, R. Determination of hydrogen in 6H–SiC epitaxial layers by the 15N nuclear reaction analysis technique. Nucl. Instrum. Methods Phys. Res. B 1999, 155, 132–136. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Ito, S.; Wilde, M.; Fukutani, K.; Hirozawa, I.; Koganezawa, T. A hydrogen storage layer on the surface of silicon nitride films. Appl. Phys. Lett. 2008, 92, 192115. [Google Scholar] [CrossRef]
- Bernard, E.; Khodja, H.; Chêne, J.; Pégourié, B.; Martin, C.; Pardanaud, C. Simultaneous deuterium implantation and ion beam microanalyses in CFC NB31: Understanding the in-bulk migration. J. Nucl. Mater. 2013, 438, S975–S978. [Google Scholar] [CrossRef]
- Kulisch, W.; Sasaki, T.; Rossi, F.; Popov, C.; Sippel, C.; Grambole, D. Hydrogen incorporation in ultrananocrystalline diamond/amorphous carbon films. Phys. Status Solidi (Rapid Res. Lett.) 2008, 2, 77–79. [Google Scholar] [CrossRef]
Figure 1.
Hydrogen solubility in various solids and melts. Note that temperature is expressed in Kelvin. Adapted according to [19]. Copyright (2004) The Japan Society of Applied Physics.
Figure 1.
Hydrogen solubility in various solids and melts. Note that temperature is expressed in Kelvin. Adapted according to [19]. Copyright (2004) The Japan Society of Applied Physics.

Figure 2.
Hydrogen analysis by IGF with TC detection (a) and IR detection (b).

Figure 3.
TDS plots of Ti–6Al–4V alloy, hydrogenated for 12 and 48 h, using a constant heating rate of 5 °C/min. Reprinted from [25]. Copyright 2007, with permission from Elsevier.
Figure 3.
TDS plots of Ti–6Al–4V alloy, hydrogenated for 12 and 48 h, using a constant heating rate of 5 °C/min. Reprinted from [25]. Copyright 2007, with permission from Elsevier.

Figure 4.
Hydrogen depth profiles of a-Si:H samples determined by GDOES (a) and NRA (b), see also Section 3.2.2. The higher was the deposition temperature, the lower is hydrogen concentration in the coating and the greater is its thickness [37]. Ordinate scale in GDOES: raw data, intensities of the H I line at 121.567 nm relative to the intensity of the Ar II line at 415.29 nm. Ordinate scale in NRA: absolute hydrogen concentration. Reproduced from Ref. [37] with a courtesy of Dr. Jonathan Steffens (University of Konstanz).
Figure 4.
Hydrogen depth profiles of a-Si:H samples determined by GDOES (a) and NRA (b), see also Section 3.2.2. The higher was the deposition temperature, the lower is hydrogen concentration in the coating and the greater is its thickness [37]. Ordinate scale in GDOES: raw data, intensities of the H I line at 121.567 nm relative to the intensity of the Ar II line at 415.29 nm. Ordinate scale in NRA: absolute hydrogen concentration. Reproduced from Ref. [37] with a courtesy of Dr. Jonathan Steffens (University of Konstanz).

Figure 5.
GDMS depth profile of an amorphous silicon solar cell. Reproduced with a courtesy of Prof. Rosario Pereiro (University of Oviedo).
Figure 5.
GDMS depth profile of an amorphous silicon solar cell. Reproduced with a courtesy of Prof. Rosario Pereiro (University of Oviedo).

Figure 6.
GDOES calibration curve of the Mo I line at 418.832 nm for analysis of molybdenum in various Ni alloys. Magenta points: raw data; red points: sputter rate-corrected and line interference-corrected data. Abscissa: line intensities; ordinate: plain (cE,M) and sputter rate-corrected (cE,M.qM) concentrations. Line interferences from other alloying elements (Fe, Ti, Nb, and W in this case) add an unwanted signal (light emission) that must be subtracted. This correction shifts the experimental points in this diagram to the left, while sputter rate correction shifts them in the vertical direction. Wavelength resolution was Δλ ≈ 0.07 nm. The slope of the trendline depicted is the reciprocal of the emission yield of Mo: 1/RMo,418.832 nm. Reproduced from [39] with permission from The Royal Society of Chemistry.
Figure 6.
GDOES calibration curve of the Mo I line at 418.832 nm for analysis of molybdenum in various Ni alloys. Magenta points: raw data; red points: sputter rate-corrected and line interference-corrected data. Abscissa: line intensities; ordinate: plain (cE,M) and sputter rate-corrected (cE,M.qM) concentrations. Line interferences from other alloying elements (Fe, Ti, Nb, and W in this case) add an unwanted signal (light emission) that must be subtracted. This correction shifts the experimental points in this diagram to the left, while sputter rate correction shifts them in the vertical direction. Wavelength resolution was Δλ ≈ 0.07 nm. The slope of the trendline depicted is the reciprocal of the emission yield of Mo: 1/RMo,418.832 nm. Reproduced from [39] with permission from The Royal Society of Chemistry.

Figure 7.
Fully resolved LIBS spectrum of Hα and Dα lines, from a fossil sample painted on the surface with heavy water. The spectrum was measured under helium gas at 5 torr. Reprinted with permission from [72]. Copyright 2006 American Chemical Society.
Figure 7.
Fully resolved LIBS spectrum of Hα and Dα lines, from a fossil sample painted on the surface with heavy water. The spectrum was measured under helium gas at 5 torr. Reprinted with permission from [72]. Copyright 2006 American Chemical Society.

Figure 8.
SIMS depth profile of niobium oxide on niobium after ion implantation of 1H, 2D, and 18O. Adapted according to [84] with permission. Copyright 2016 John Wiley & Sons.
Figure 8.
SIMS depth profile of niobium oxide on niobium after ion implantation of 1H, 2D, and 18O. Adapted according to [84] with permission. Copyright 2016 John Wiley & Sons.

Figure 9.
RBS (a) and ERDA (b) spectra of amorphous carbon film on silicon. Impurity levels are H ≈ 1 at.%, Ar ≈ 1 at.%, and O ≈ 2 at.%. In the RBS spectrum, hydrogen is not visible and the peak of carbon (Mr = 12) is superposed on a background caused by silicon (Mr = 28). Reprinted from [97] with the permission of AIP Publishing.
Figure 9.
RBS (a) and ERDA (b) spectra of amorphous carbon film on silicon. Impurity levels are H ≈ 1 at.%, Ar ≈ 1 at.%, and O ≈ 2 at.%. In the RBS spectrum, hydrogen is not visible and the peak of carbon (Mr = 12) is superposed on a background caused by silicon (Mr = 28). Reprinted from [97] with the permission of AIP Publishing.


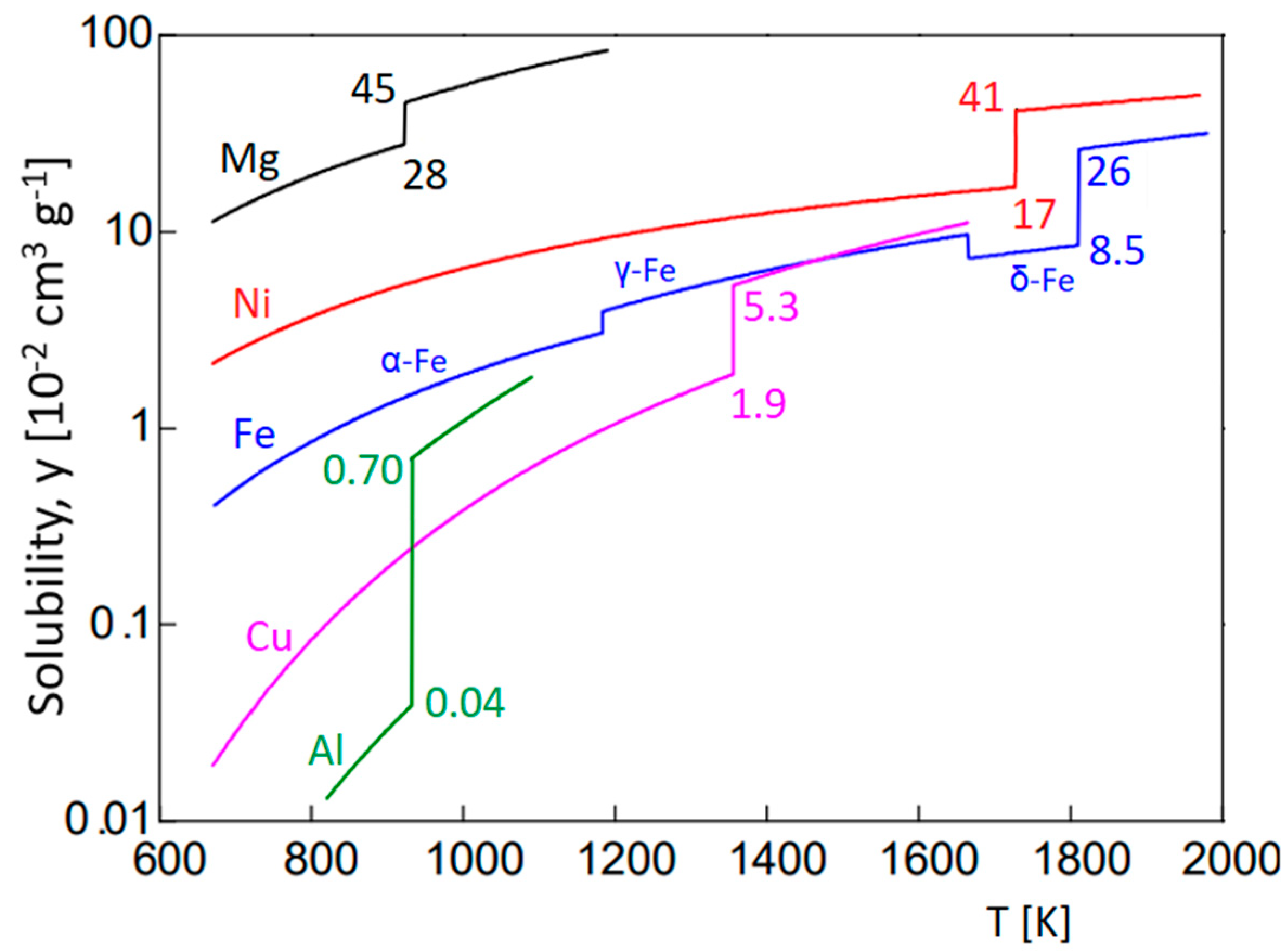{kind=link}
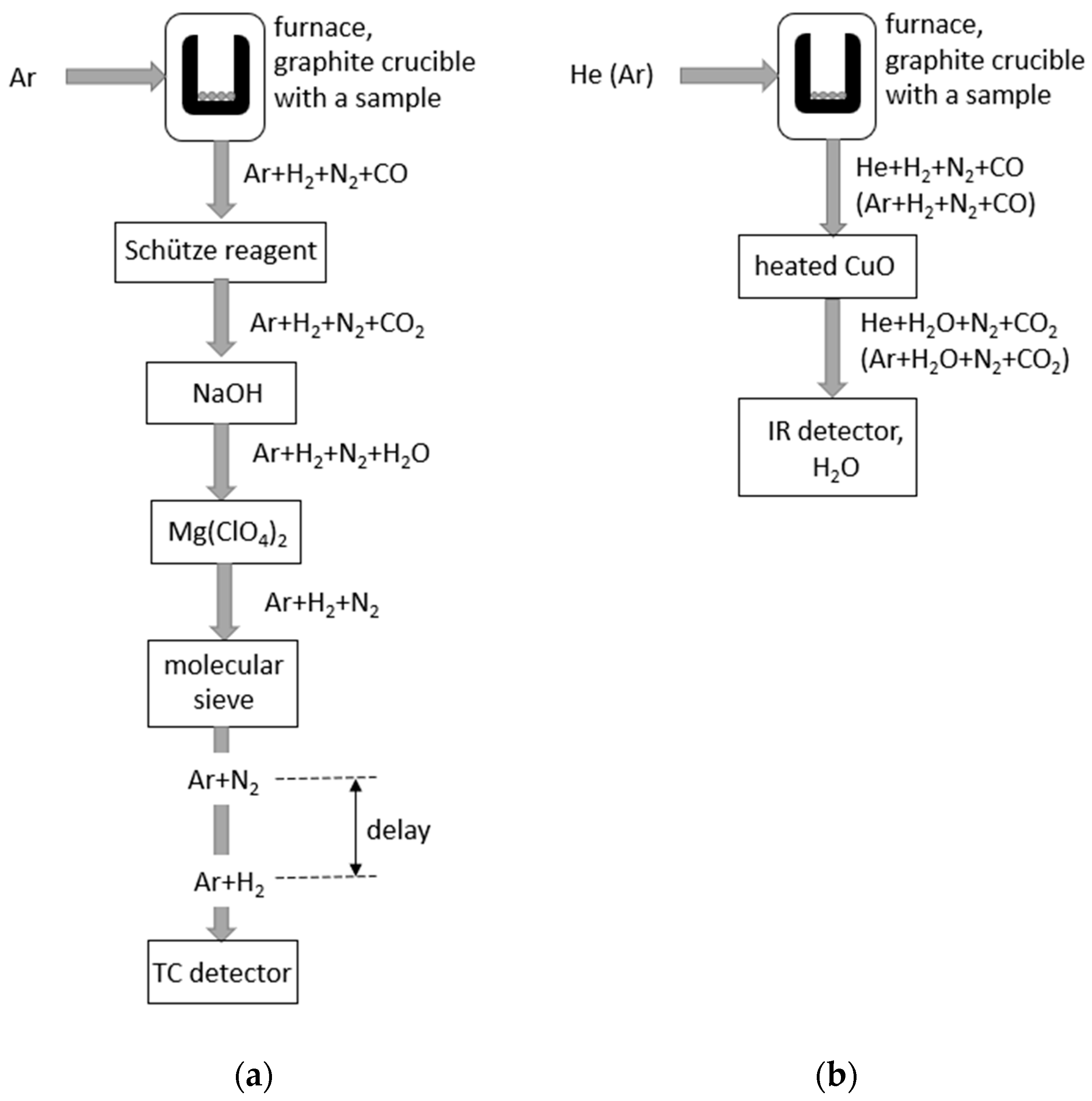{kind=link}
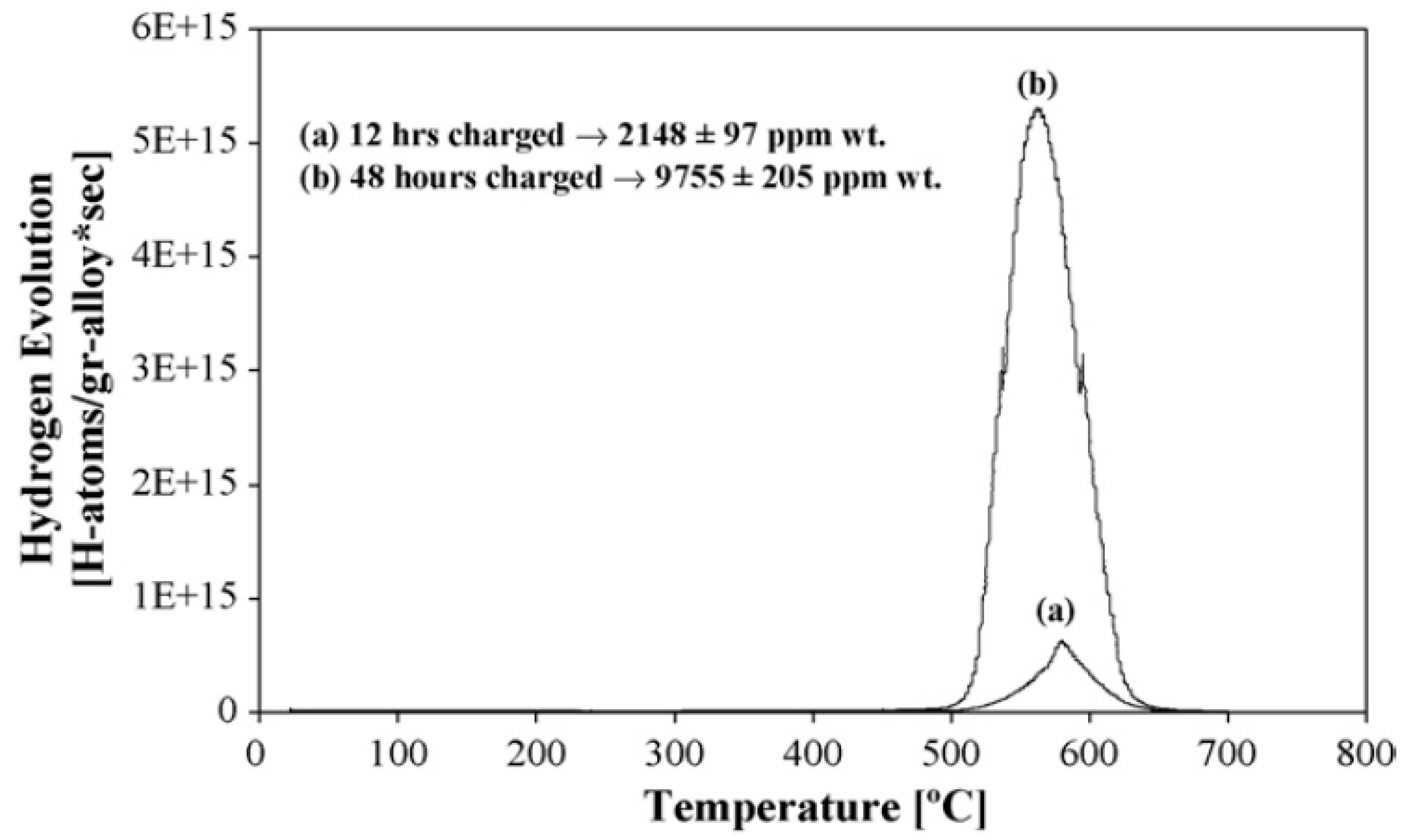{kind=link}
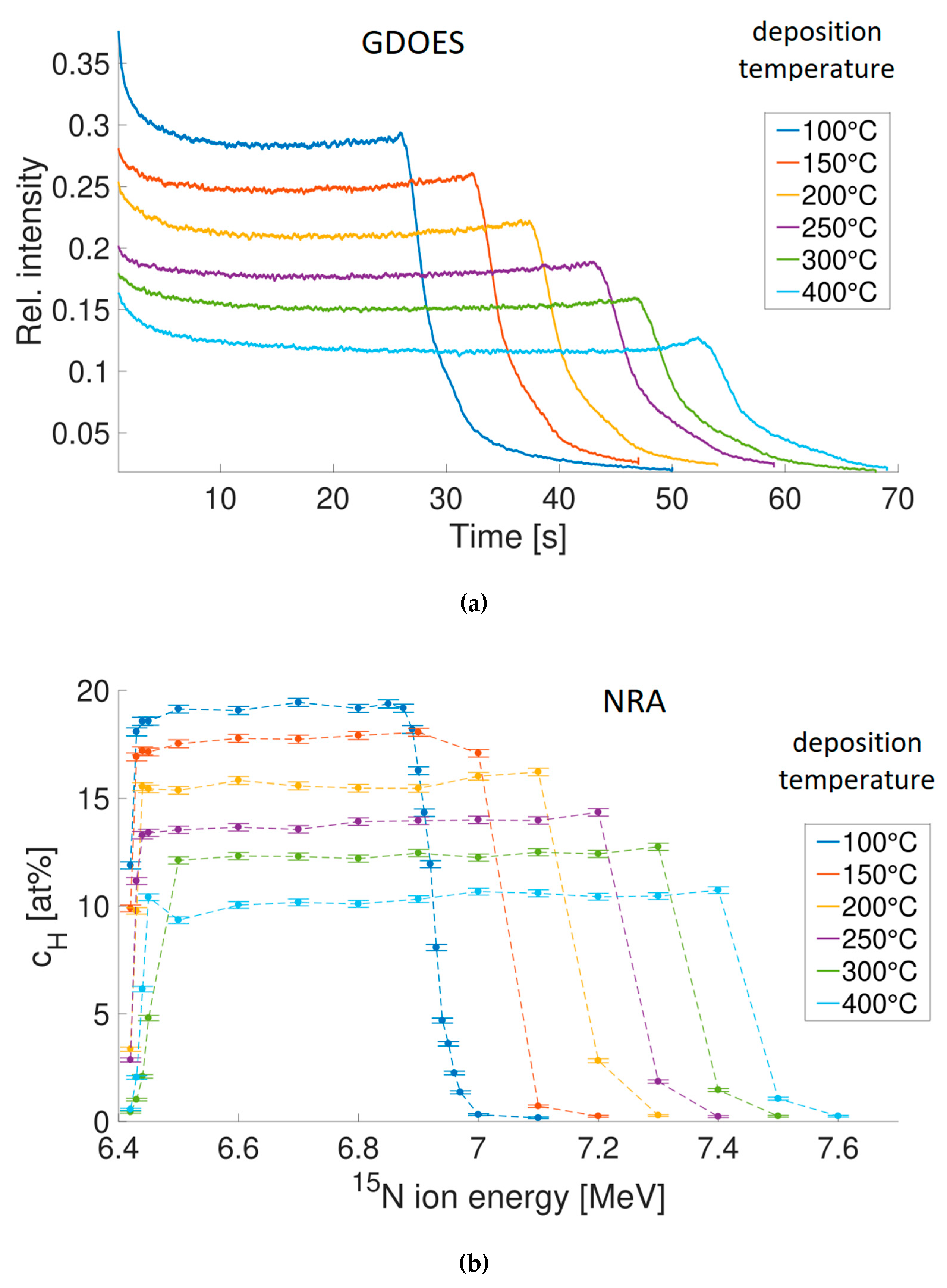{kind=link}
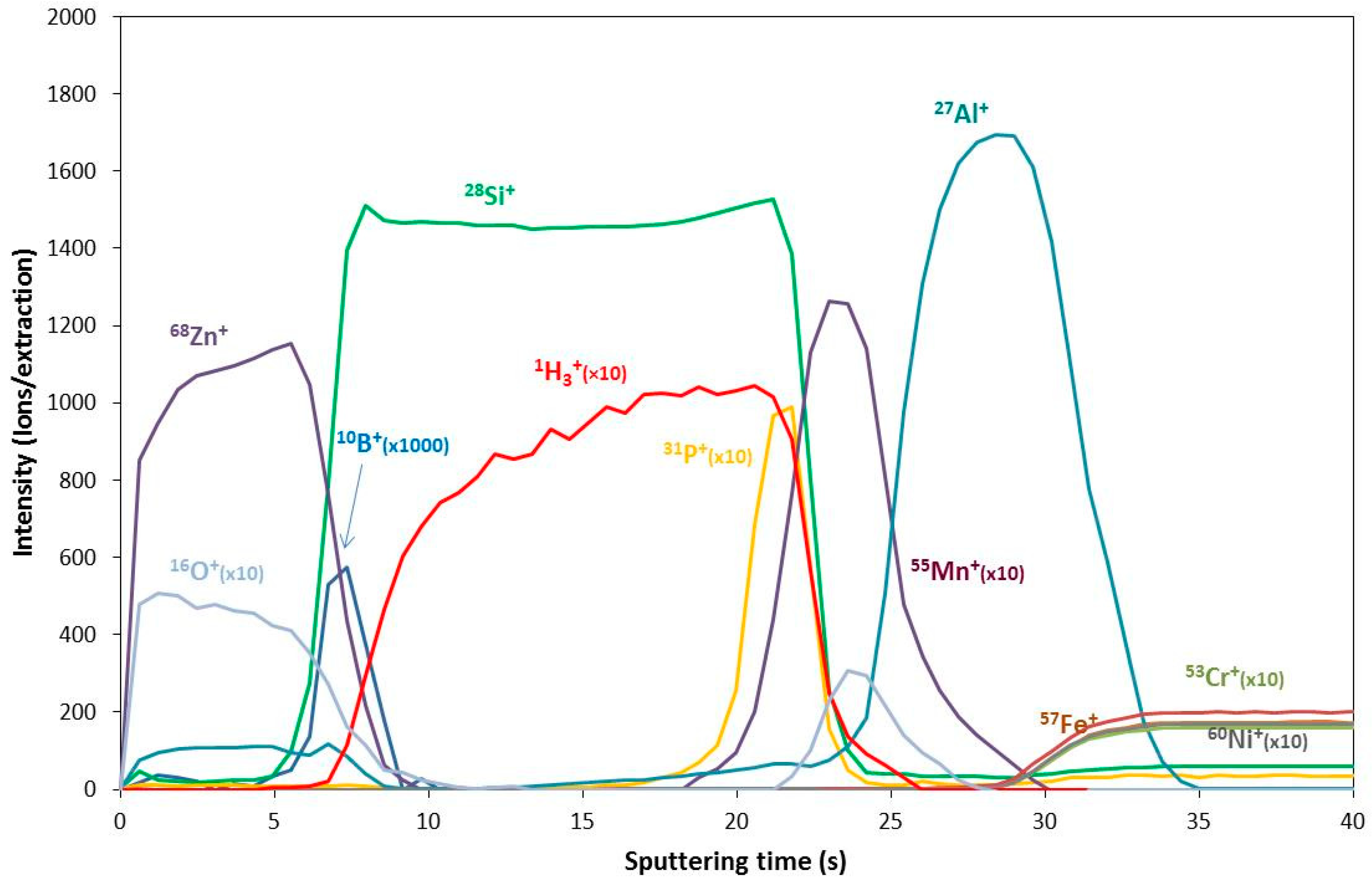{kind=link}
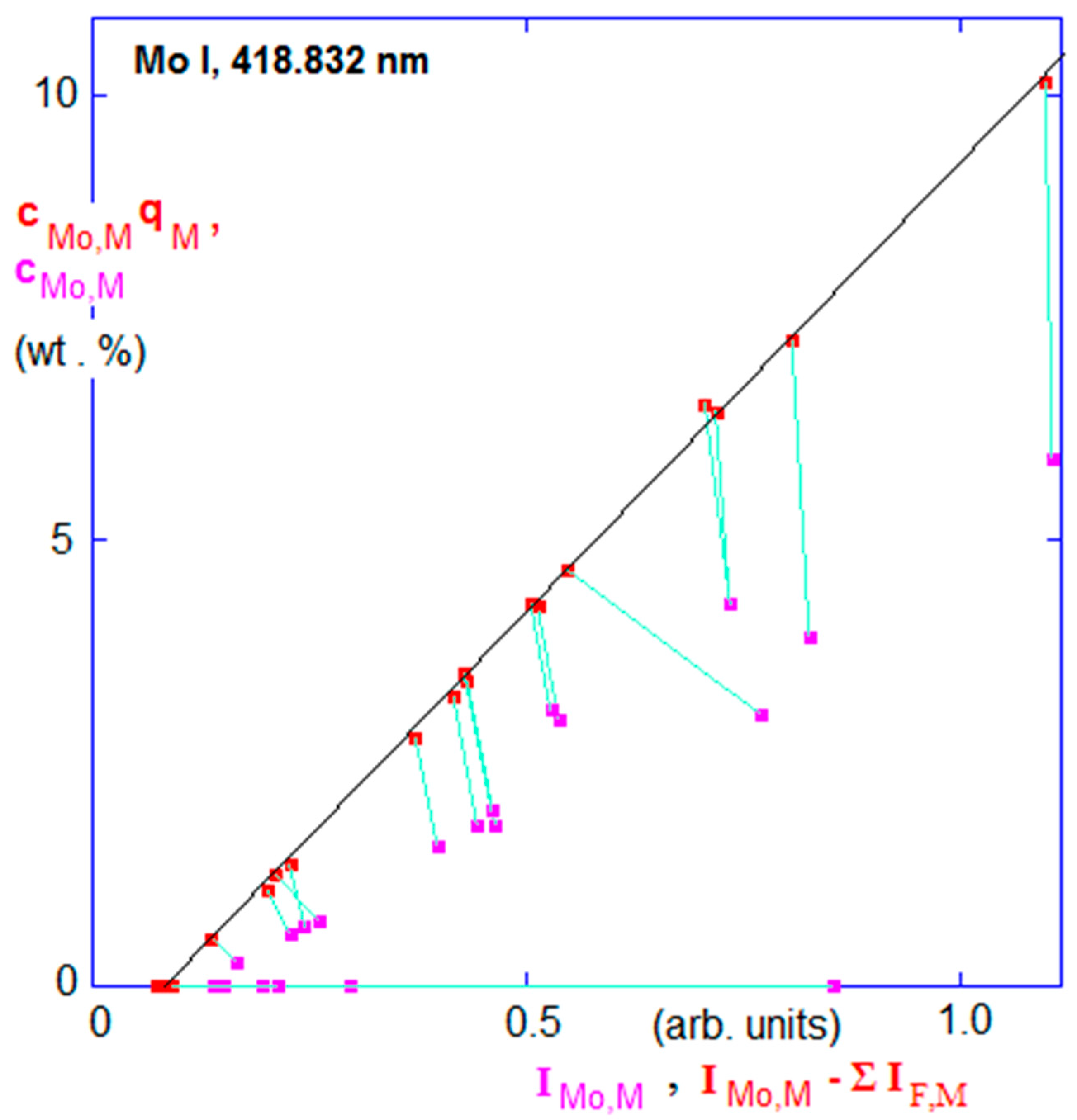{kind=link}
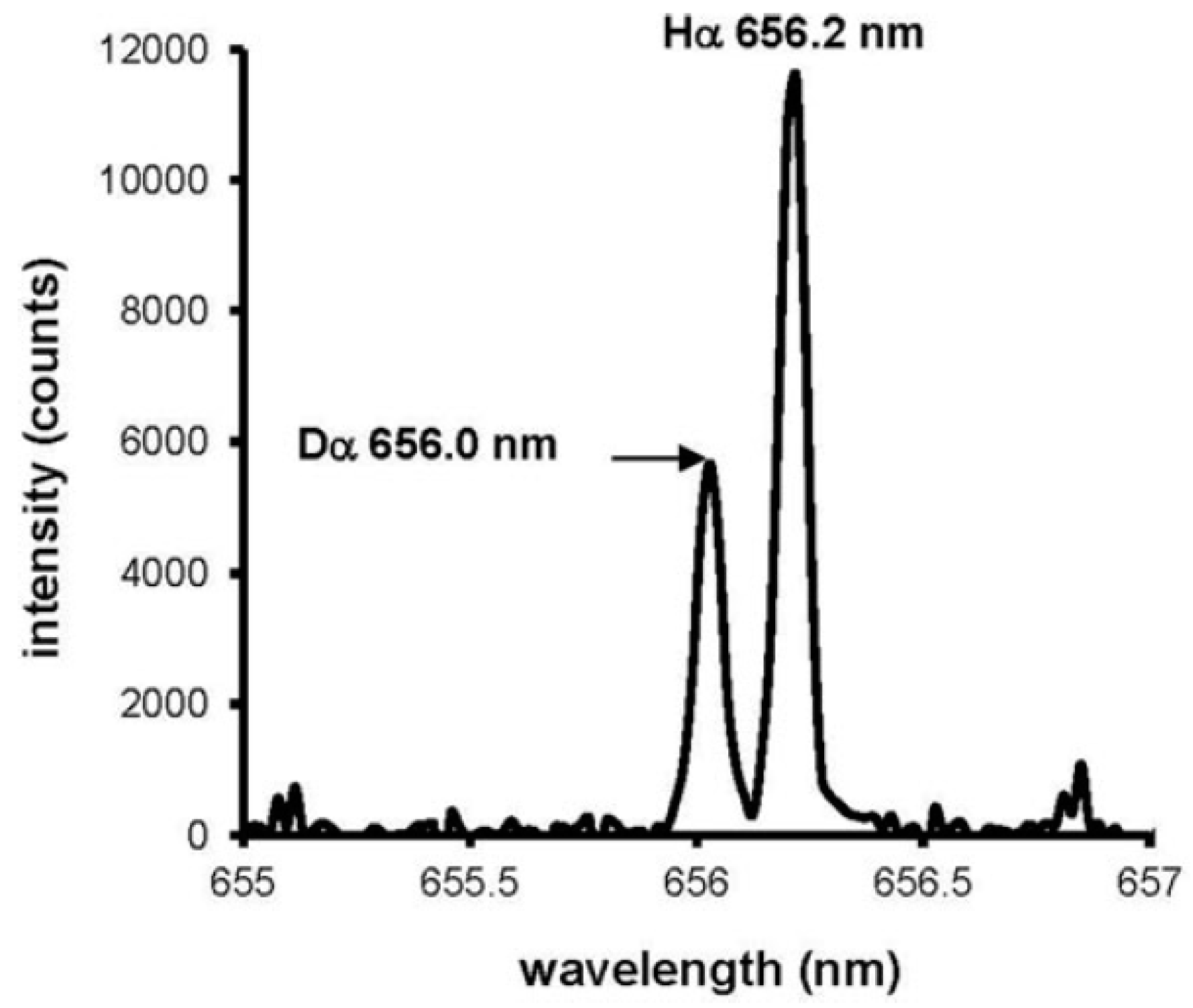{kind=link}
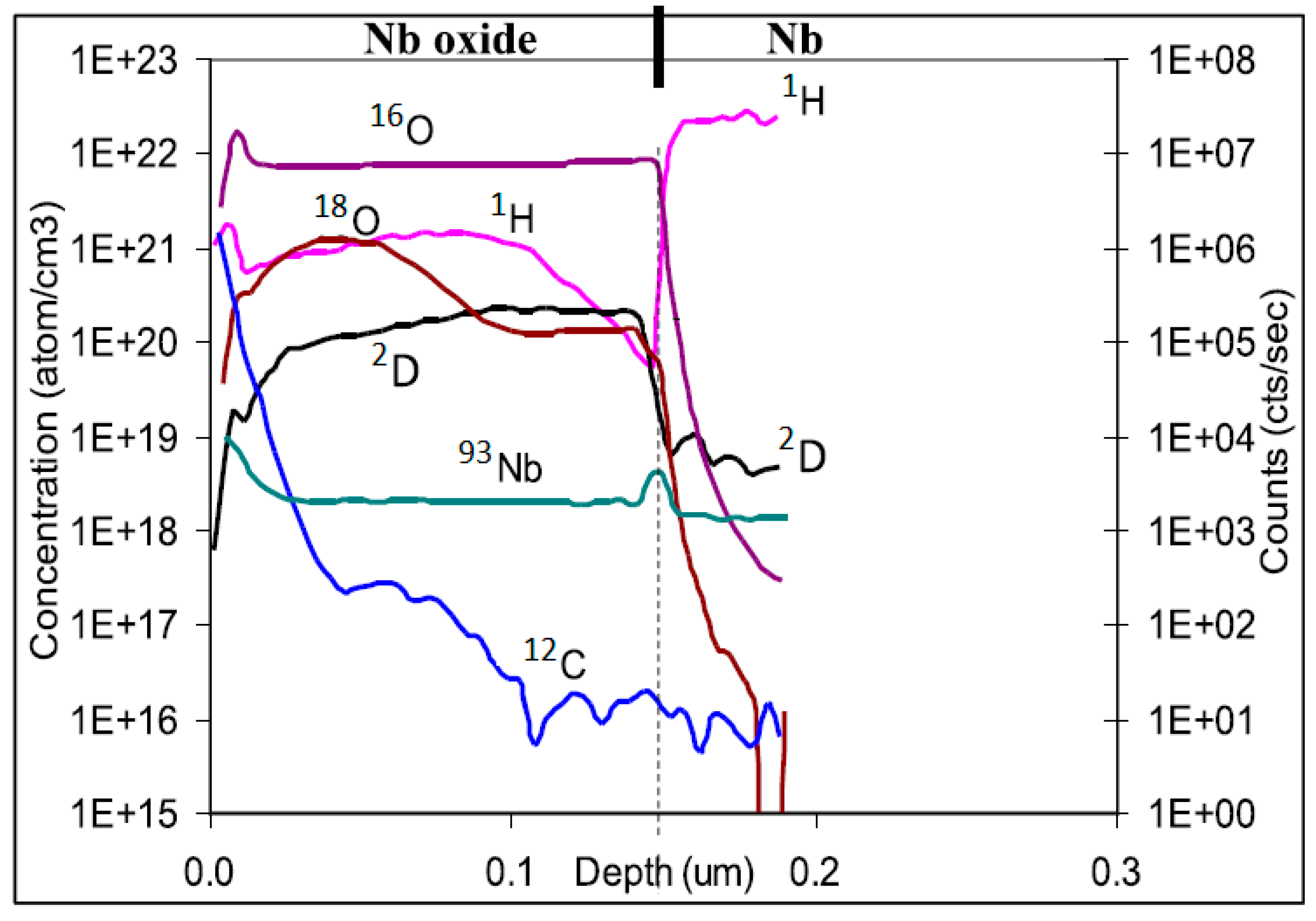{kind=link}
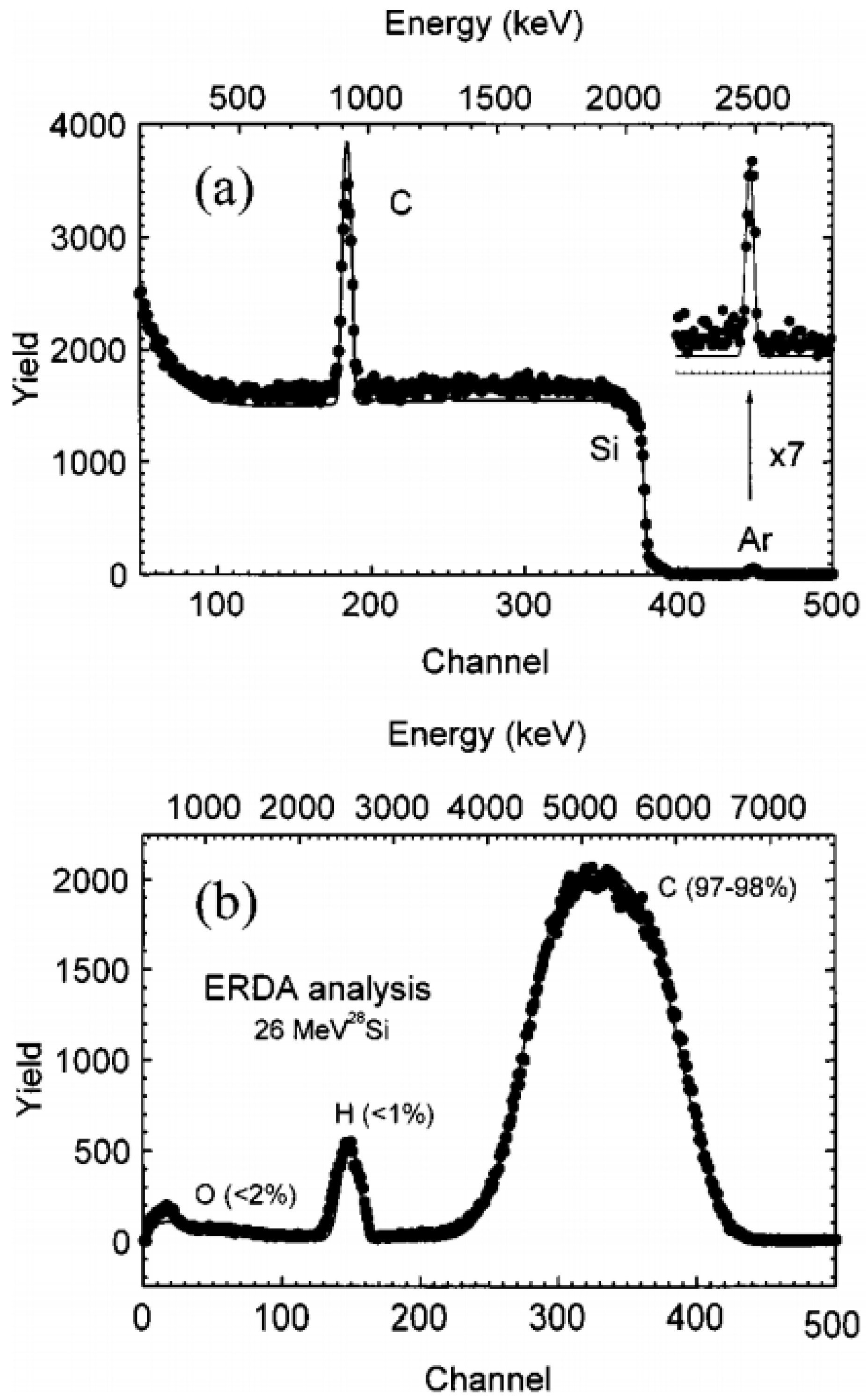{kind=link}
Table 1.
Reactions and reagents used to modify the gas composition between the furnace and the detector in the IGF analysis.
Table 1.
Reactions and reagents used to modify the gas composition between the furnace and the detector in the IGF analysis.
| Process | Reagent | Trade Name | |
|---|---|---|---|
| Conversion of CO to CO2 | CO → CO2 | Heated CuO | |
| I2O5 + H2SO4 | Schütze reagent 1 | ||
| Removal of CO2 | CO2 + 2NaOH → Na2CO3 + H2O | NaOH | |
| Conversion of H2 to H2O | H2 → H2O | Heated CuO | |
| Removal of H2O | H2O → Mg(ClO4)2·xH2O | Mg(ClO4)2 | Anhydrone™ |
1 Schütze reagent is used to convert CO into CO2, at room temperature. It leaves hydrogen, H2, unaffected. Heated CuO, on the other hand, oxidizes both CO to CO2 and H2 to H2O. Heated CuO also converts methane, CH4, into CO2 and H2O, and it is preferably used, together with IR detection, if there is a chance of CH4 being released.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weiss, Z. Analysis of Hydrogen in Inorganic Materials and Coatings: A Critical Review. Hydrogen 2021, 2, 225-245. https://doi.org/10.3390/hydrogen2020012
AMA Style
Weiss Z. Analysis of Hydrogen in Inorganic Materials and Coatings: A Critical Review. Hydrogen. 2021; 2(2):225-245. https://doi.org/10.3390/hydrogen2020012
Chicago/Turabian StyleWeiss, Zdeněk. 2021. "Analysis of Hydrogen in Inorganic Materials and Coatings: A Critical Review" Hydrogen 2, no. 2: 225-245. https://doi.org/10.3390/hydrogen2020012