Controllable H2 Generation by Formic Acid Decomposition on a Novel Pd/Templated Carbon Catalyst


Abstract
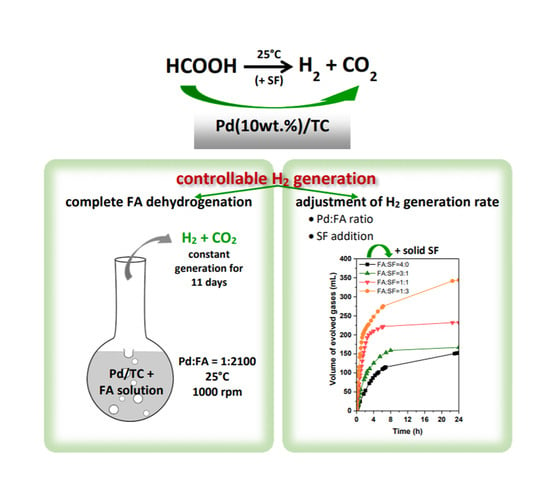
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Catalyst Preparation
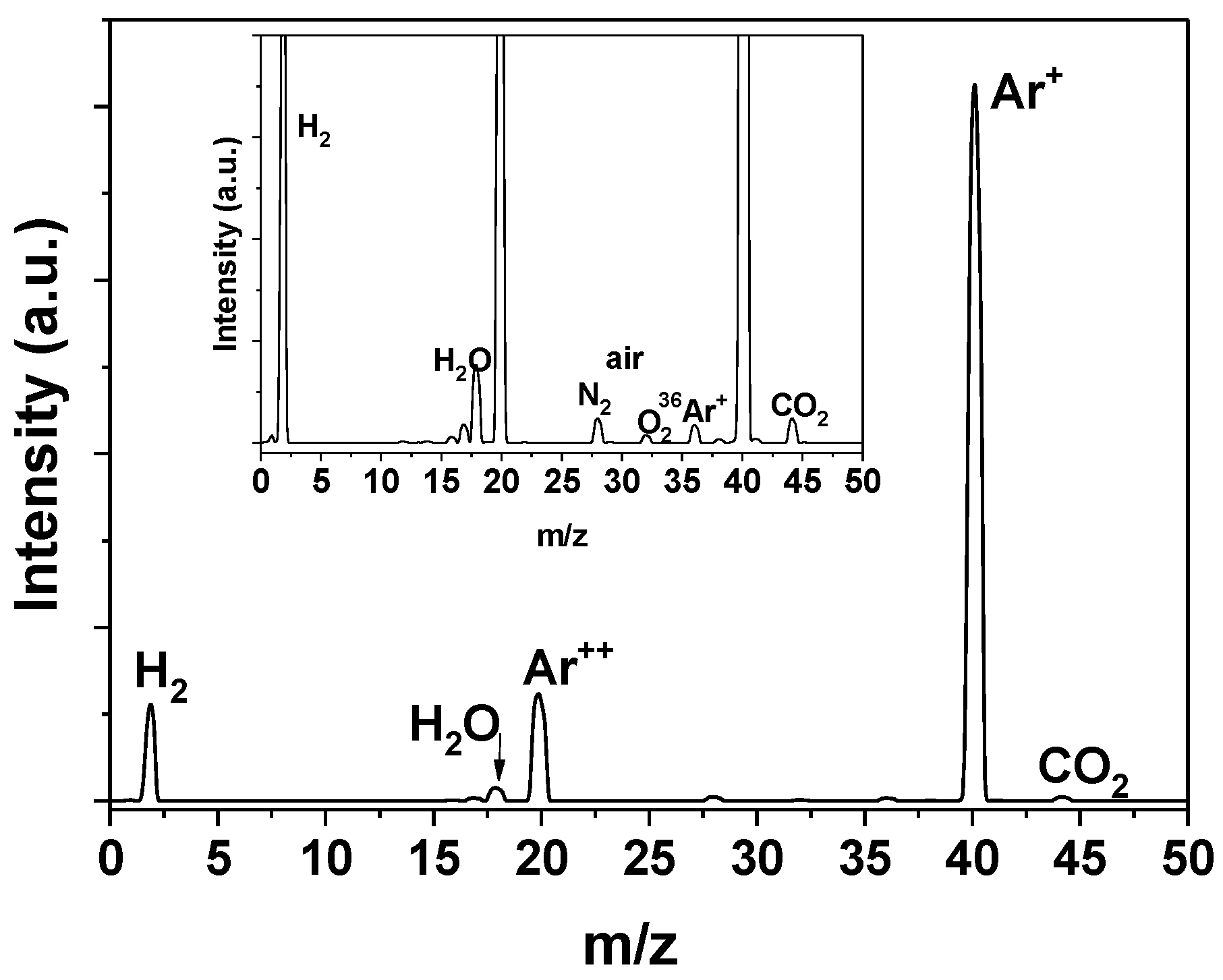
2.3. Catalyst Characterization
2.4. Catalytic Activity Tests
3. Results and Discussion
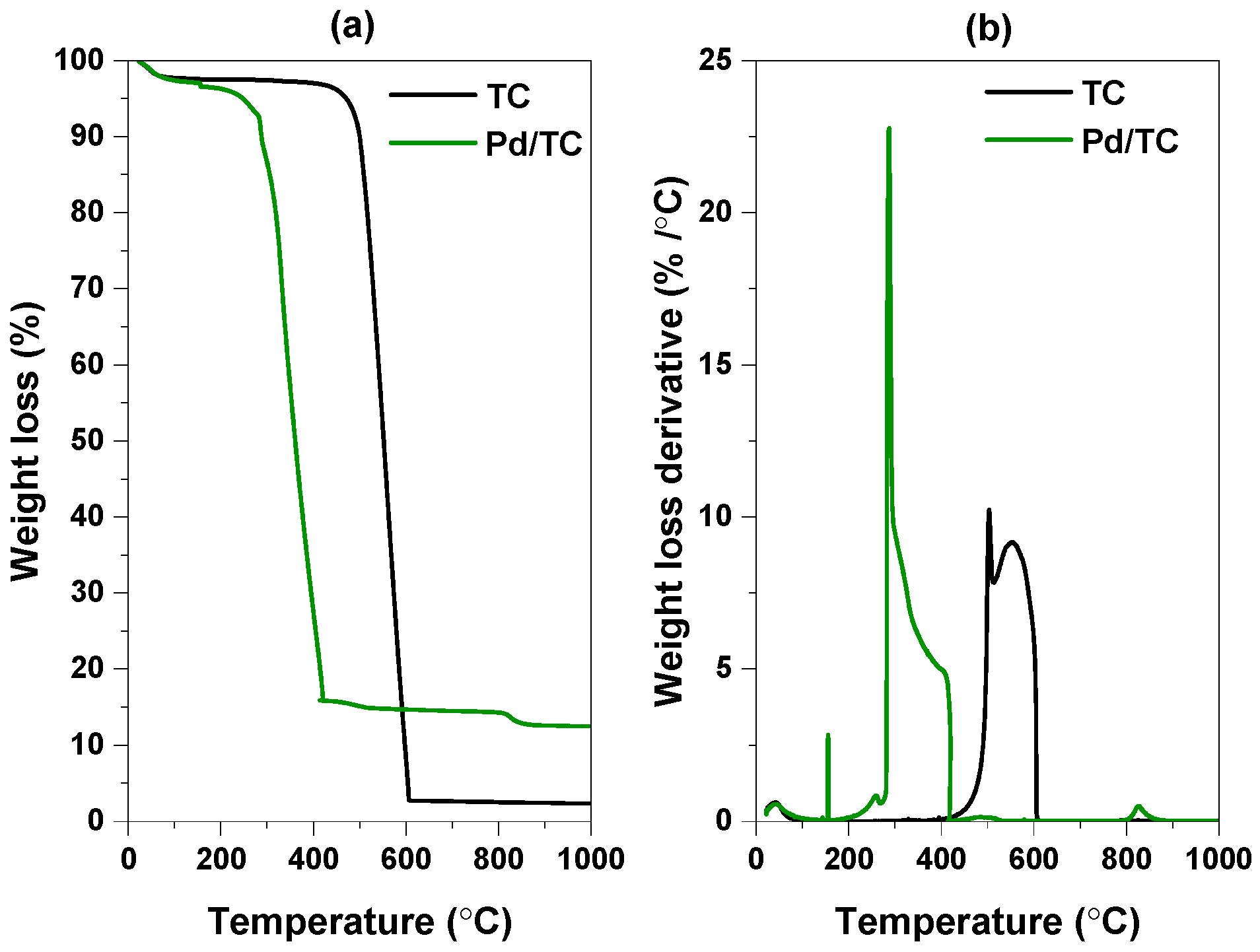
3.1. Characterization Results
3.2. Catalytic Activity Results
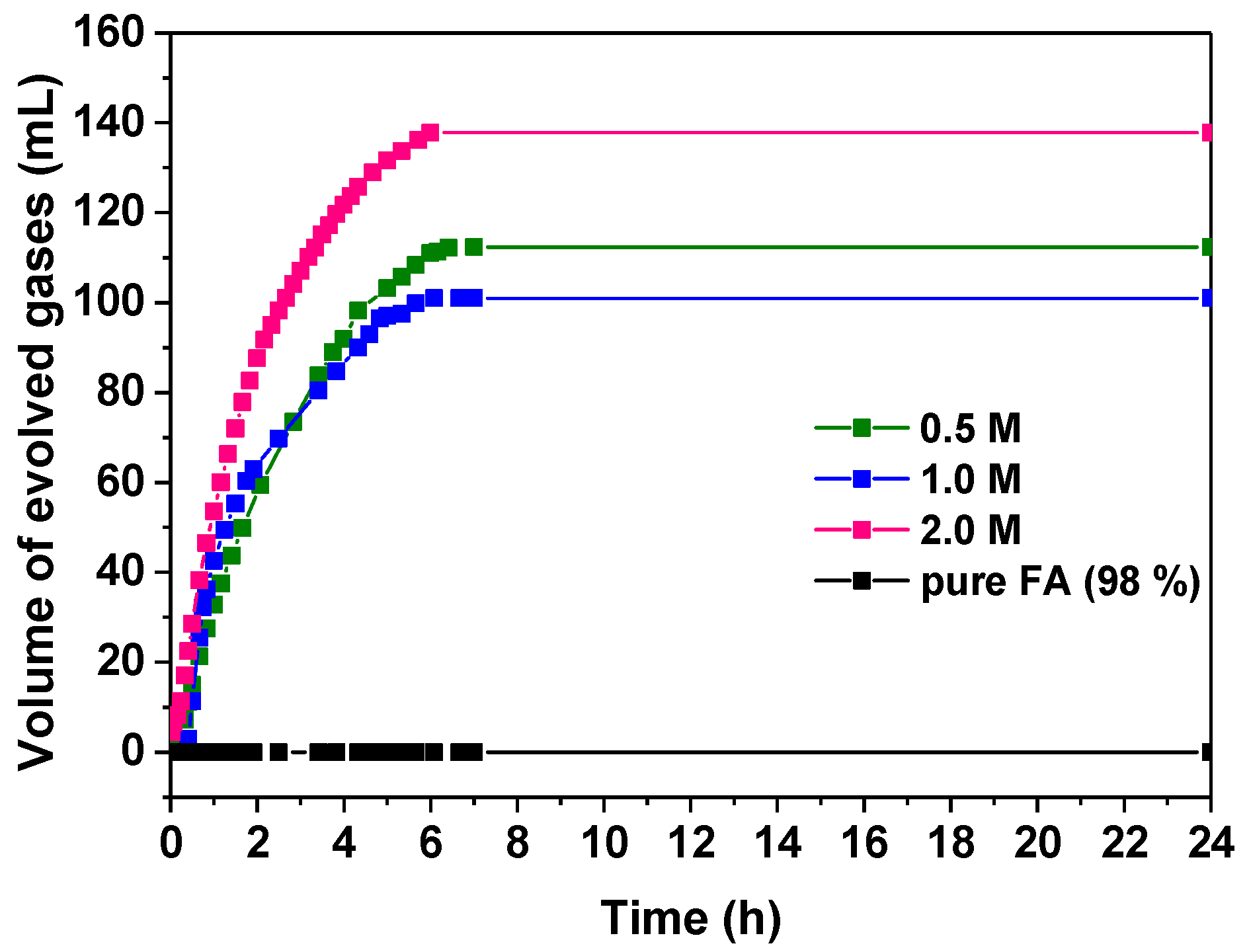
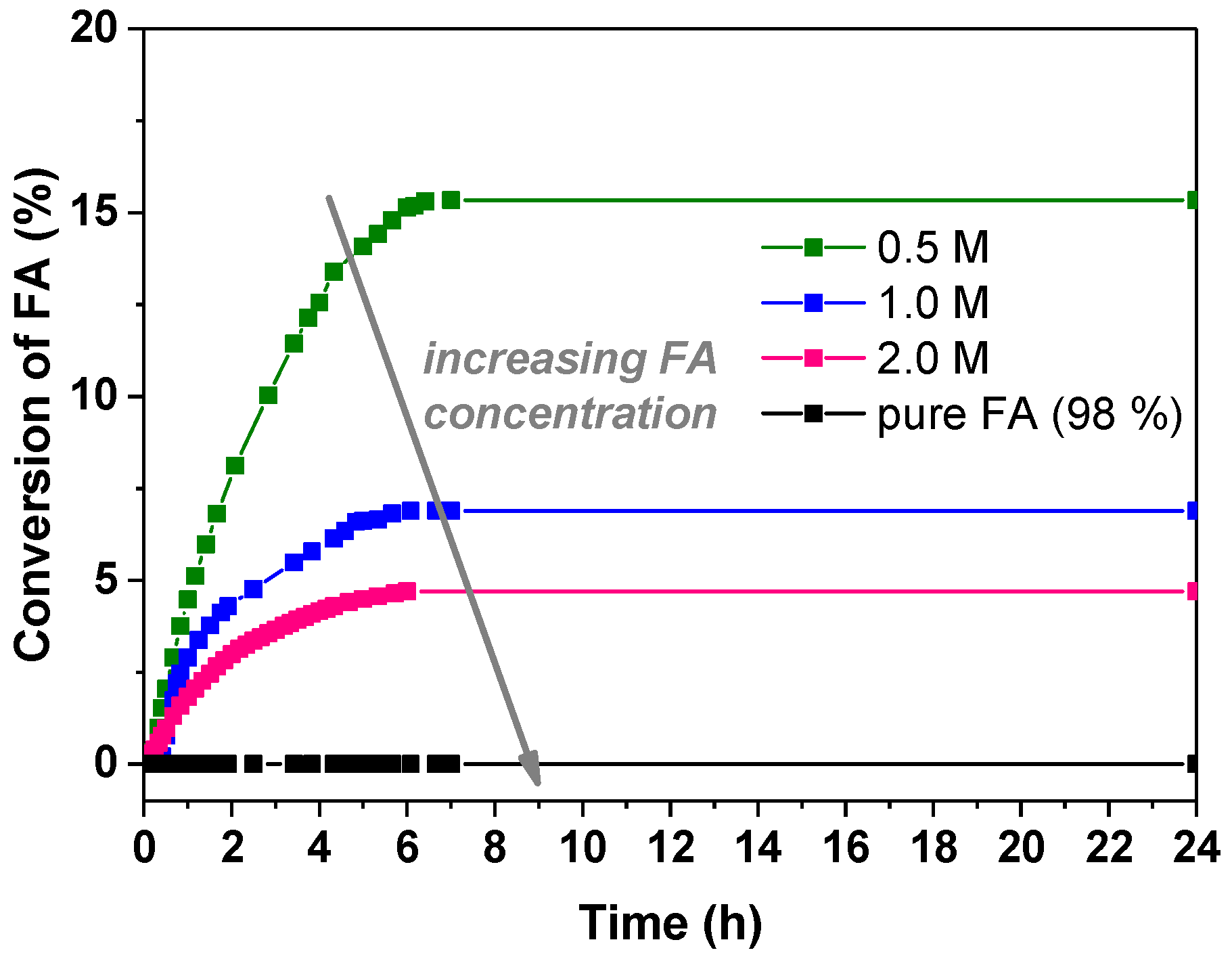
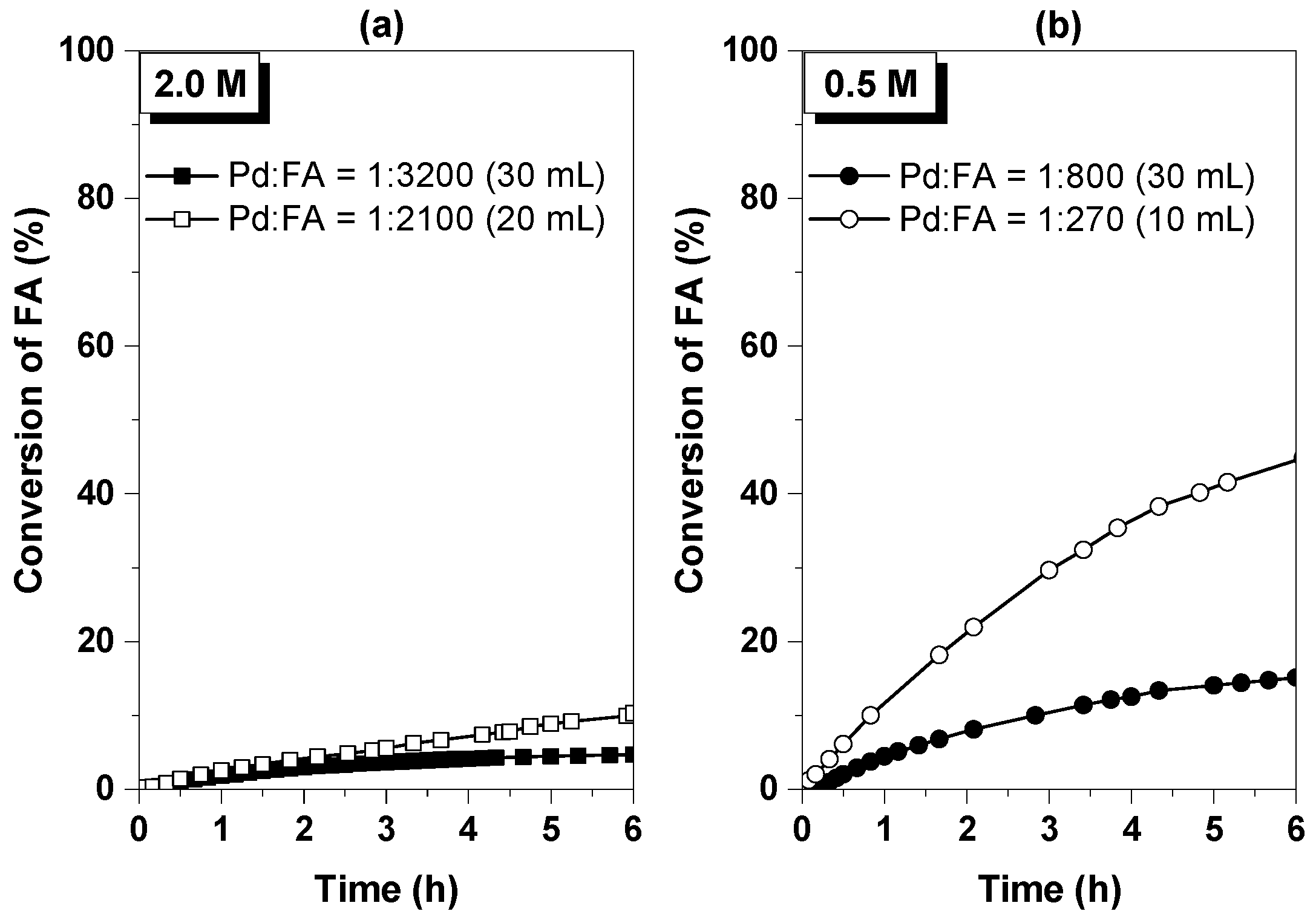
3.2.1. Influence of the Formic Acid Concentration
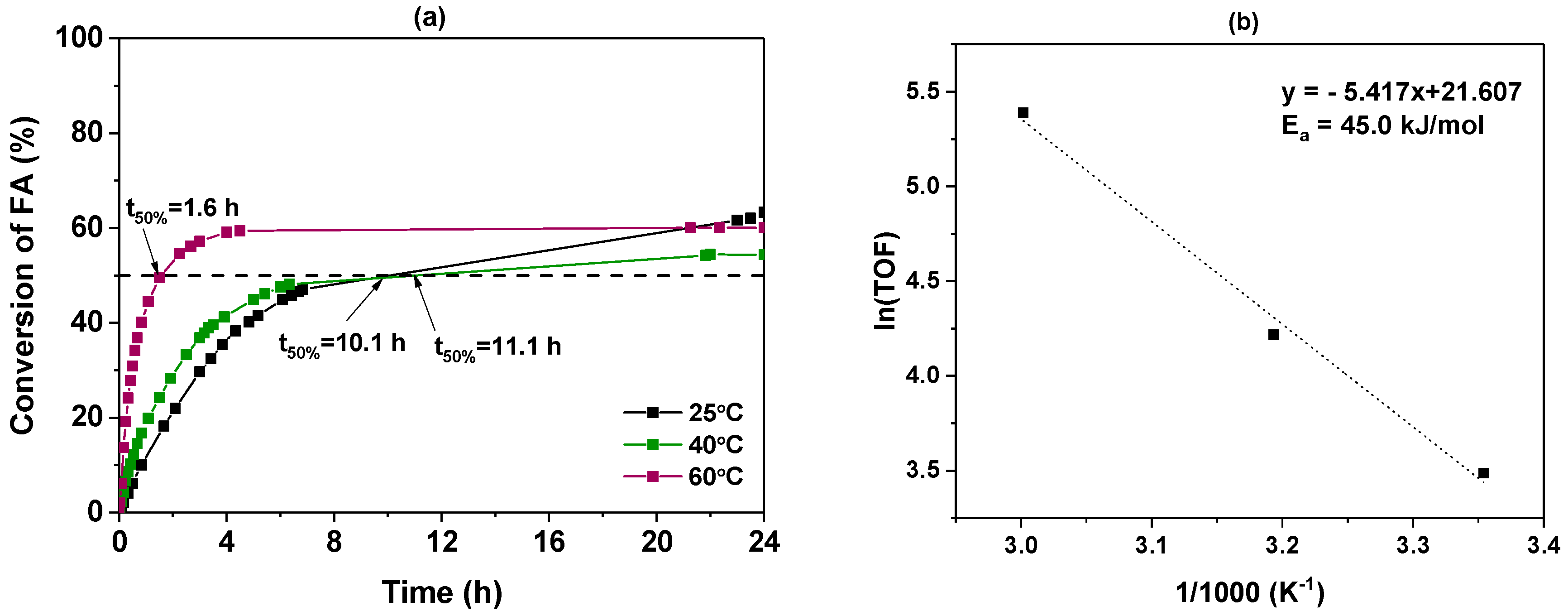
3.2.2. Influence of Temperature
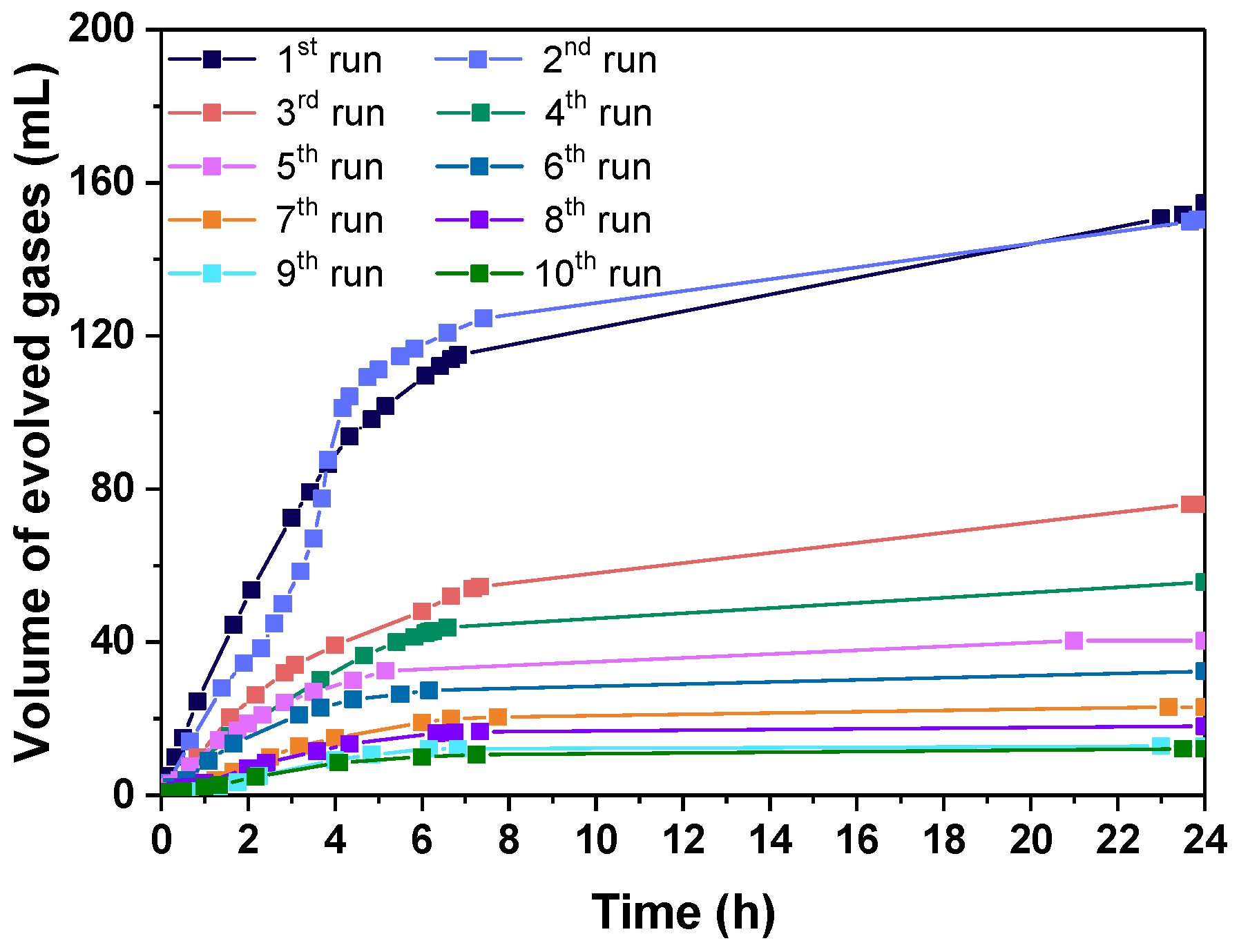
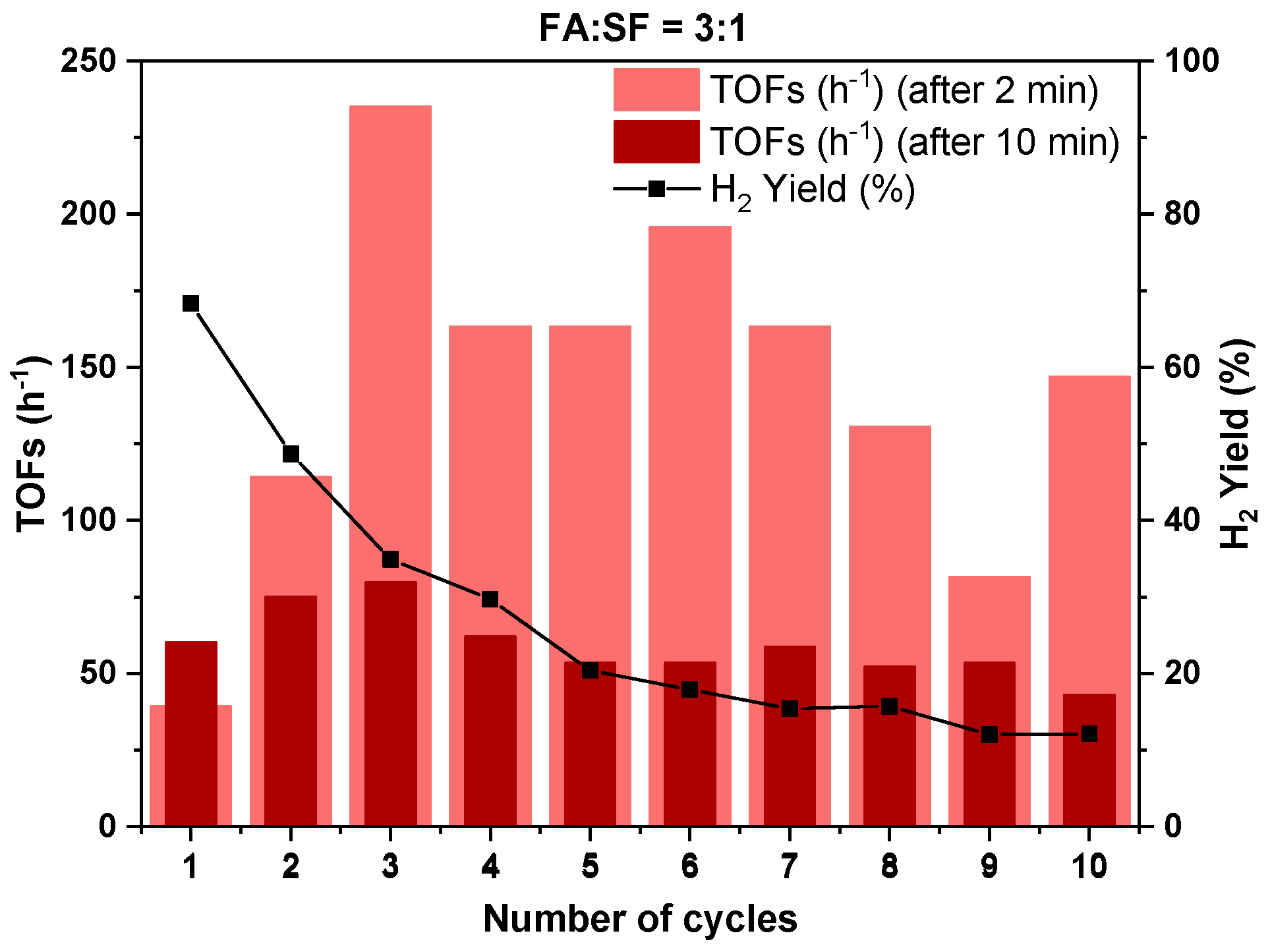
3.2.3. Catalyst Life Time and Complete FA Dehydrogenation
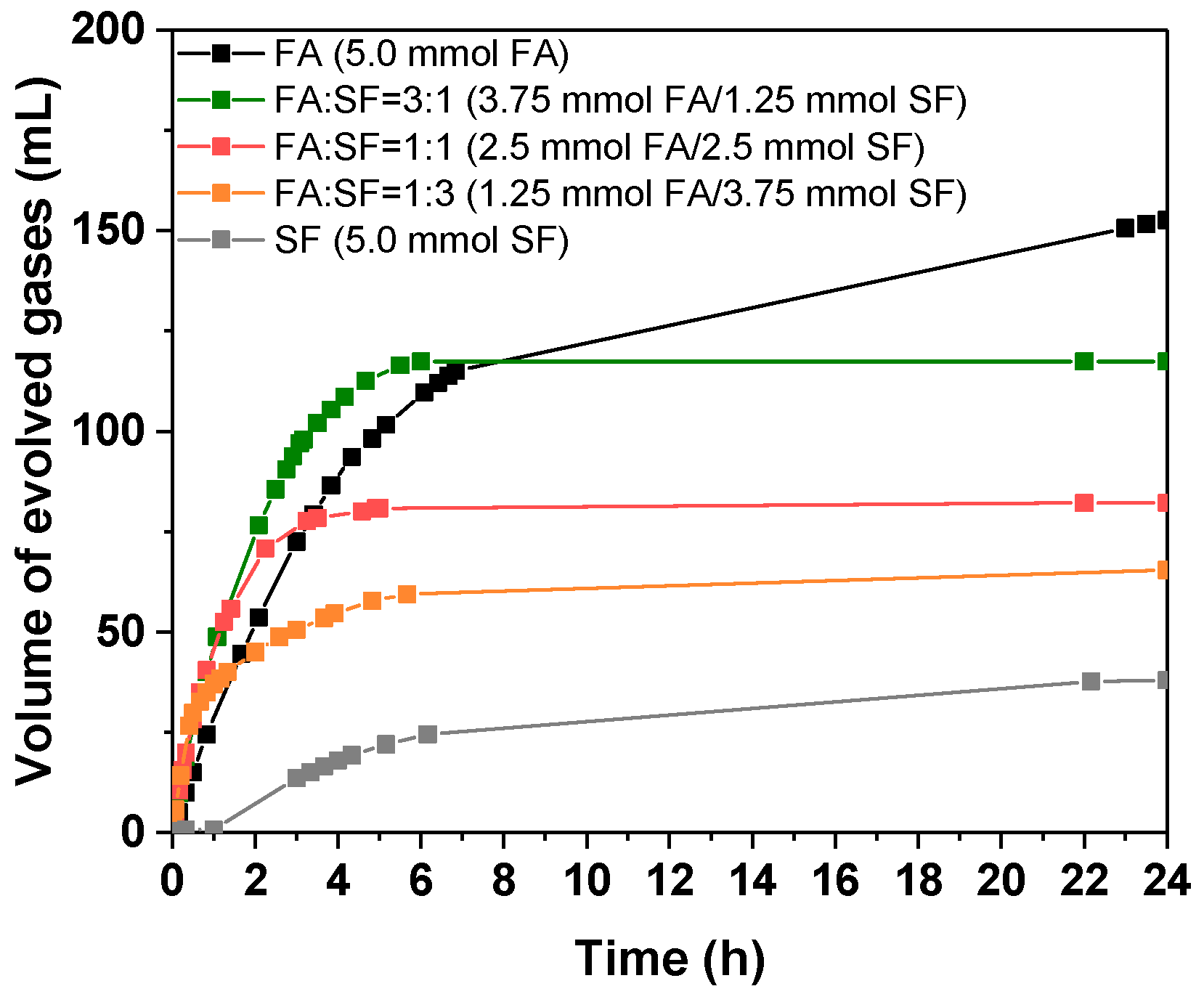
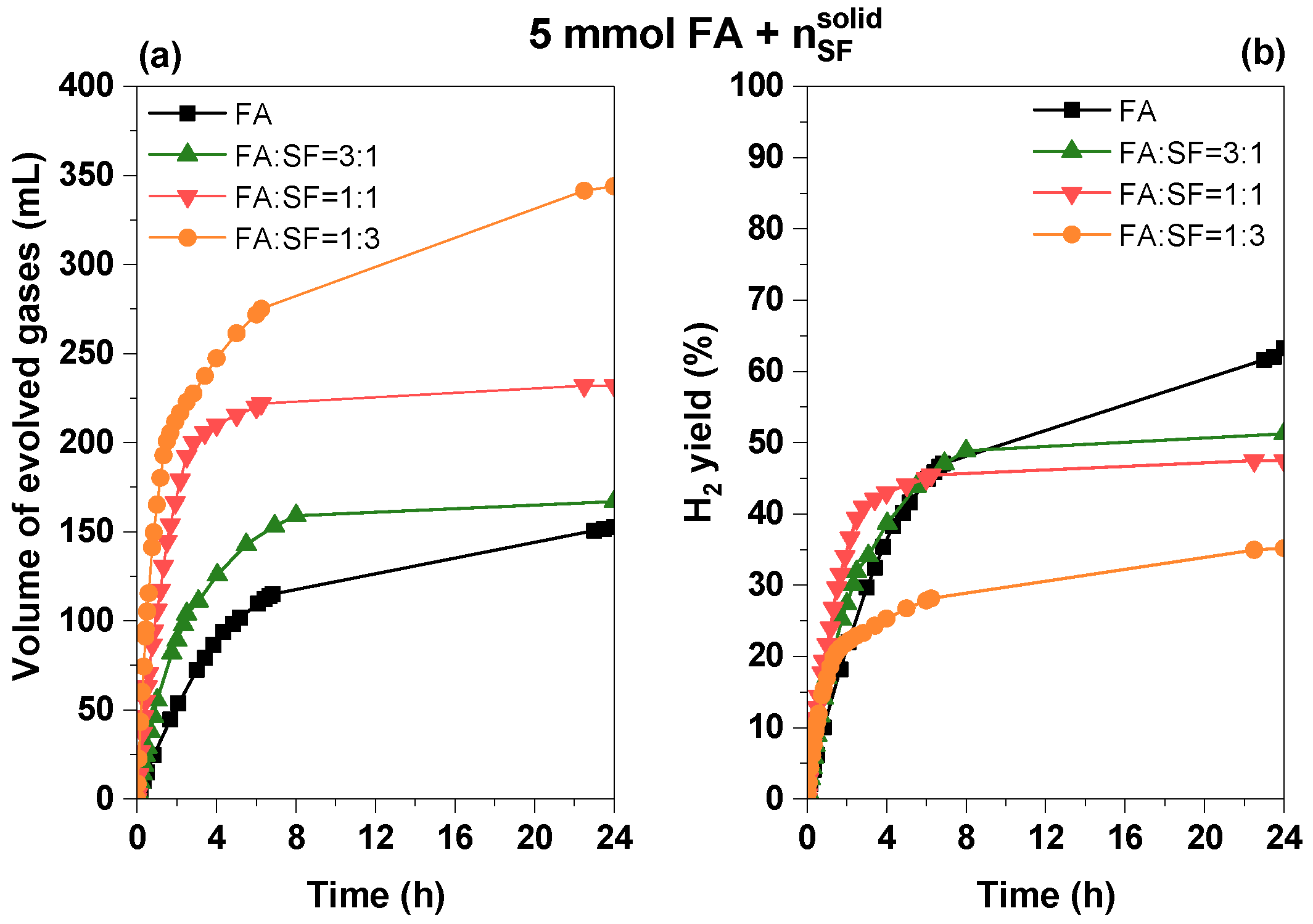
3.2.4. Influence of Sodium Formate Addition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source – recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Wang, H.; Ping, Y.; Jiang, Q. Pd/C Synthesized with Citric Acid: An Efficient Catalyst for Hydrogen Generation from Formic Acid/Sodium Formate. Sci. Rep. 2012, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Ryu, J.; Kim, J.Y.; Nam, S.-W.; Han, J.H.; Lim, T.-H.; Gautam, S.; Chae, K.H.; Yoon, C.W. Carbon dioxide mediated, reversible chemical hydrogen storage using a Pd nanocatalyst supported on mesoporous graphitic carbon nitride. J. Mater. Chem. A 2014, 2, 9490–9495. [Google Scholar] [CrossRef]
- Jeon, H.-J.; Chung, Y.-M. Hydrogen production from formic acid dehydrogenation over Pd/C catalysts: Effect of metal and support properties on the catalytic performance. Appl. Catal. B Environ. 2017, 210, 212–222. [Google Scholar] [CrossRef]
- Wang, X.; Qi, G.-W.; Tan, C.-H.; Li, Y.-P.; Guo, J.; Pang, X.-J.; Zhang, S.-Y. Pd/C nanocatalyst with high turnover frequency for hydrogen generation from the formic acid-formate mixtures. Int. J. Hydrogen Energy 2014, 39, 837–843. [Google Scholar] [CrossRef]
- Tedsree, K.; Li, T.; Jones, S.; Chan, C.W.A.; Yu, K.M.K.; Bagot, P.A.J.; Marquis, E.A.; Smith, G.D.W.; Tsang, S.C.E. Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat. Nanotechnol. 2011, 6, 302–307. [Google Scholar] [CrossRef]
- Jena, P.J. Materials for Hydrogen Storage: Past, Present, and Future. Phys. Chem. Lett. 2011, 2, 206–211. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Strategies for Hydrogen Storage in Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2005, 44, 4670–4679. [Google Scholar] [CrossRef]
- Blanita, G.; Coldea, I.; Misan, I.; Lupu, D. Hydrogen cryo-adsorption by hexagonal prism monoliths of MIL-101. Int. J. Hydrogen Energy 2014, 39, 17040–17046. [Google Scholar] [CrossRef]
- Ardelean, O.; Blanita, G.; Borodi, G.; Lazar, M.D.; Misan, I.; Coldea, I.; Lupu, D. Volumetric hydrogen adsorption capacity of densified MIL-101 monoliths. Int. J. Hydrogen Energy 2013, 38, 7046–7055. [Google Scholar] [CrossRef]
- Jiang, H.-L.; Singh, S.K.; Yan, J.-M.; Zhang, X.-B.; Xu, Q. Liquid-Phase Chemical Hydrogen Storage: Catalytic Hydrogen Generation under Ambient Conditions. ChemSusChem 2010, 3, 541–549. [Google Scholar] [CrossRef]
- Zhou, J.-P.; Zhang, J.; Dai, X.-H.; Wang, X.; Zhang, S.-Y. Formic acid-ammonium formate mixture: A new system with extremely high dehydrogenation activity and capacity. Int. J. Hydrogen Energy 2016, 41, 22059–22066. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Ping, Y.; Wang, H.-L.; Zheng, W.-T.; Jiang, Q. An Efficient CoAuPd/C Catalyst for Hydrogen Generation from Formic Acid at Room Temperature. Angew. Chem. Int. Ed. 2013, 52, 4406–4409. [Google Scholar] [CrossRef]
- Akbayrak, S.; Tonbul, Y.; Ozkar, S. Nanoceria supported palladium(0) nanoparticles: Superb catalyst in dehydrogenation of formic acid at room temperature. Appl. Catal. B Environ. 2017, 206, 384–392. [Google Scholar] [CrossRef]
- Enthaler, S.; von Langermann, J.; Schmidt, T. Carbon dioxide and formic acid—the couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010, 3, 1207–1217. [Google Scholar] [CrossRef]
- Yang, L.; Hua, X.; Su, J.; Luo, W.; Chen, S.; Cheng, G. Highly efficient hydrogen generation from formic acid-sodium formate over monodisperse AgPd nanoparticles at room temperature. Appl. Catal. B Environ. 2015, 168–169, 423–428. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Ping, Y.; Yan, J.-M.; Wang, H.; Jiang, Q. Hydrogen generation from formic acid decomposition at room temperature using a NiAuPd alloy nanocatalyst. Int. J. Hydrogen Energy 2014, 39, 4850–4856. [Google Scholar] [CrossRef]
- Karatas, Y.; Bulut, A.; Yurderi, M.; Ertas, I.E.; Alal, O.; Gulcan, M.; Celebi, M.; Kivrak, H.; Kaya, M.; Zahmakiran, M. PdAu-MnOx nanoparticles supported on amine-functionalized SiO2 for the room temperature dehydrogenation of formic acid in the absence of additives. Appl. Catal. B Environ. 2016, 180, 586–595. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Beloshapkin, S.; Ross, J.R.H. Hydrogen from formic acid decomposition over Pd and Au catalysts. Catal. Today 2010, 154, 7–12. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, S.; Chul, H.; Heui, D.J. Mechanistic insights on aqueous formic acid dehydrogenation over Pd/C catalyst for efficient hydrogen production. J. Catal. 2020, 389, 506–516. [Google Scholar] [CrossRef]
- Grad, O.; Mihet, M.; Coros, M.; Dan, M.; Lazar, M.D.; Blanita, G. Reduced graphene oxide modified with noble metal nanoparticles for formic acid dehydrogenation. Catal. Today 2020, in press. [Google Scholar] [CrossRef]
- Zacharska, M.; Chuvilin, A.L.; Kriventsov, V.V.; Beloshapkin, S.; Estrada, M.; Simakov, A.; Bulushev, D.A. Support effect for nanosized Au catalysts in hydrogen production from formic acid decomposition. Catal. Sci. Technol. 2016, 6, 6853–6860. [Google Scholar] [CrossRef] [Green Version]
- Bi, Q.-Y.; Du, X.-L.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Efficient Subnanometric Gold-Catalyzed Hydrogen Generation via Formic Acid Decomposition under Ambient Conditions. J. Am. Chem. Soc. 2012, 134, 8926–8933. [Google Scholar] [CrossRef] [PubMed]
- Gazsi, A.; Bansagi, T.; Solymosi, F.J. Decomposition and Reforming of Formic Acid on Supported Au Catalysts: Production of CO-Free H2. Phys. Chem. C 2011, 115, 15459–15466. [Google Scholar] [CrossRef] [Green Version]
- Grad, O.; Mihet, M.; Dan, M.; Blanita, G.; Berghian-Grosan, C.; Lazar, M.D. Au/reduced graphene oxide composites: Eco-friendly preparation method and catalytic applications for formic acid dehydrogenation. J. Mater. Sci. 2019, 54, 6991–7004. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, Y.; Xing, W.; Liu, C.; Liao, J.; Lu, T. High-quality hydrogen from the catalyzed decomposition of formic acid by Pd–Au/C and Pd–Ag/C. Chem. Commun. 2008, 30, 3540–3542. [Google Scholar] [CrossRef]
- Gu, X.; Lu, Z.-H.; Jiang, H.; Akita, T.; Xu, Q.J. Synergistic catalysis of metal-organic framework-immobilized Au-Pd nanoparticles in dehydrogenation of formic acid for chemical hydrogen storage. Am. Chem. Soc. 2011, 133, 11822–11825. [Google Scholar] [CrossRef]
- Dai, H.; Cao, N.; Yang, L.; Su, J.; Luo, W.; Cheng, G.J. AgPd nanoparticles supported on MIL-101 as high performance catalysts for catalytic dehydrogenation of formic acid. Mater. Chem. A 2014, 2, 11060–11064. [Google Scholar] [CrossRef]
- Hu, C.; Ting, S.-W.; Tsui, J.; Chan, K.-Y. Formic acid dehydrogenation over PtRuBiOx/C catalyst for generation of CO-free hydrogen in a continuous-flow reactor. Int. J. Hydrogen Energy 2012, 37, 6372–6380. [Google Scholar] [CrossRef]
- Doustkhah, E.; Hasani, M.; Ide, Y.; Assadi, M.H.N. Pd Nanoalloys for H2 Generation from Formic Acid. ACS Appl. Nano Mater. 2020, 3, 22–43. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.X.; Yamazaki, T.; Orikasa, H.; Kyotani, T. An easy method for the synthesis of ordered microporous carbons by the template technique. Carbon 2005, 43, 2624–2627. [Google Scholar] [CrossRef]
- Ma, Z.; Kyotani, T.; Tomita, A. Synthesis methods for preparing microporous carbons with a structural regularity of zeolite Y. Carbon 2002, 40, 2367–2374. [Google Scholar] [CrossRef]
- Doustkhah, E.; Mohtasham, H.; Farjzadeh, M.; Rostamnia, S.; Wang, Y.; Arandiyan, H.; Assadi, M.H.N. Organosiloxane tunability in mesoporous organosilica and punctuated Pd nanoparticles growth; theory and experiment. Microporous Mesoporous Mater. 2020, 293, 109832. [Google Scholar] [CrossRef]
- Ojeda, M.; Iglesia, E. Formic Acid Dehydrogenation on Au-Based Catalysts at Near- Ambient Temperatures. Angew. Chem. Int. Ed. 2009, 48, 4800–4803. [Google Scholar] [CrossRef] [Green Version]
- Rootsaert, W.M.H.; Sachtler, W.J.M. Interaction of Formic Acid Vapour with Tungsten. Z. Phys. Chem. 1960, 26, 16–26. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FA Concentration | Pd:FA Ratio | (%) | TOF (h−1) | |
|---|---|---|---|---|
| Initial (t = 2 min) | at | |||
| 0.5 M | 1:800 | 15.33 | 15.7 | 35.6 |
| 1.0 M | 1:1600 | 6.89 | 39.2 | 38.5 |
| 2.0 M | 1:3200 | 4.7 | 143.7 | 34 |
| FA 98% | 1:42,200 | 0 | 0 | 0 |
| Temperature (°C) | [a] (h) | TOF10 min [b] (h−1) | (%) |
|---|---|---|---|
| 25 | 10.1 | 32.7 | 63.3 |
| 40 | 11.1 | 67.9 | 54.4 |
| 60 | 1.6 | 218.8 | 60.1 |
| System [FA] + [SF] = 0.5 mol/L | Vgases (mL) | TOF10 min (h−1) | Φ4h (%) | Φ24h (%) |
|---|---|---|---|---|
| FA | 154.6 | 32.7 | 35.4 | 63.3 |
| FA:SF = 3:1 | 117.4 | 64.0 | 29.4 | 48.1 |
| FA:SF = 1:1 | 82.1 | 68.6 | 21.5 | 33.6 |
| FA:SF = 1:3 | 60.0 | 77.3 | 15.2 | 24.6 |
| SF | 38.0 | 3.2 | 4.9 | 15.6 |
| System (5 mmol FA + nSF)/10 mL Solution | Vgases (mL) | TOF10min (h−1) | Φ4h (%) | Φ24h (%) |
|---|---|---|---|---|
| FA | 154.6 | 32.7 | 35.4 | 63.3 |
| FA:SF = 3:1 | 166.8 | 60.1 | 38.7 | 51.3 |
| FA:SF = 1:1 | 232.0 | 173.1 | 43.0 | 47.5 |
| FA:SF = 1:3 | 369.2 | 282.1 | 25.3 | 37.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihet, M.; Dan, M.; Barbu-Tudoran, L.; Lazar, M.D.; Blanita, G. Controllable H2 Generation by Formic Acid Decomposition on a Novel Pd/Templated Carbon Catalyst. Hydrogen 2020, 1, 22-37. https://doi.org/10.3390/hydrogen1010003
Mihet M, Dan M, Barbu-Tudoran L, Lazar MD, Blanita G. Controllable H2 Generation by Formic Acid Decomposition on a Novel Pd/Templated Carbon Catalyst. Hydrogen. 2020; 1(1):22-37. https://doi.org/10.3390/hydrogen1010003
Chicago/Turabian StyleMihet, Maria, Monica Dan, Lucian Barbu-Tudoran, Mihaela D. Lazar, and Gabriela Blanita. 2020. "Controllable H2 Generation by Formic Acid Decomposition on a Novel Pd/Templated Carbon Catalyst" Hydrogen 1, no. 1: 22-37. https://doi.org/10.3390/hydrogen1010003