
A Review of Fifteen Years Developing Computational Tools to Study Protein Aggregation

 ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Tools to Study Protein Aggregation
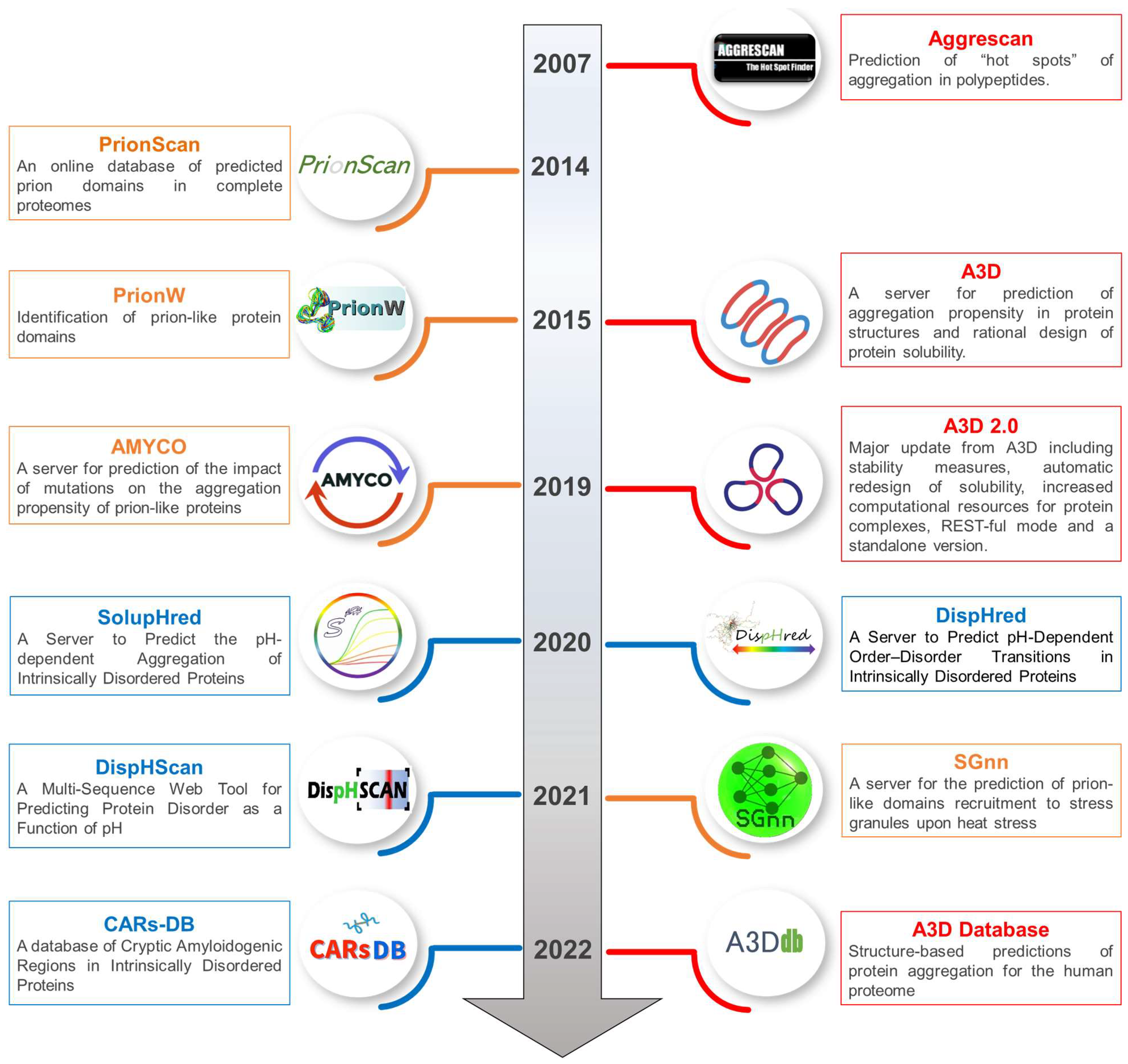
2.1. Aggrescan: Prediction of “Hot Spots” of Aggregation in Polypeptides
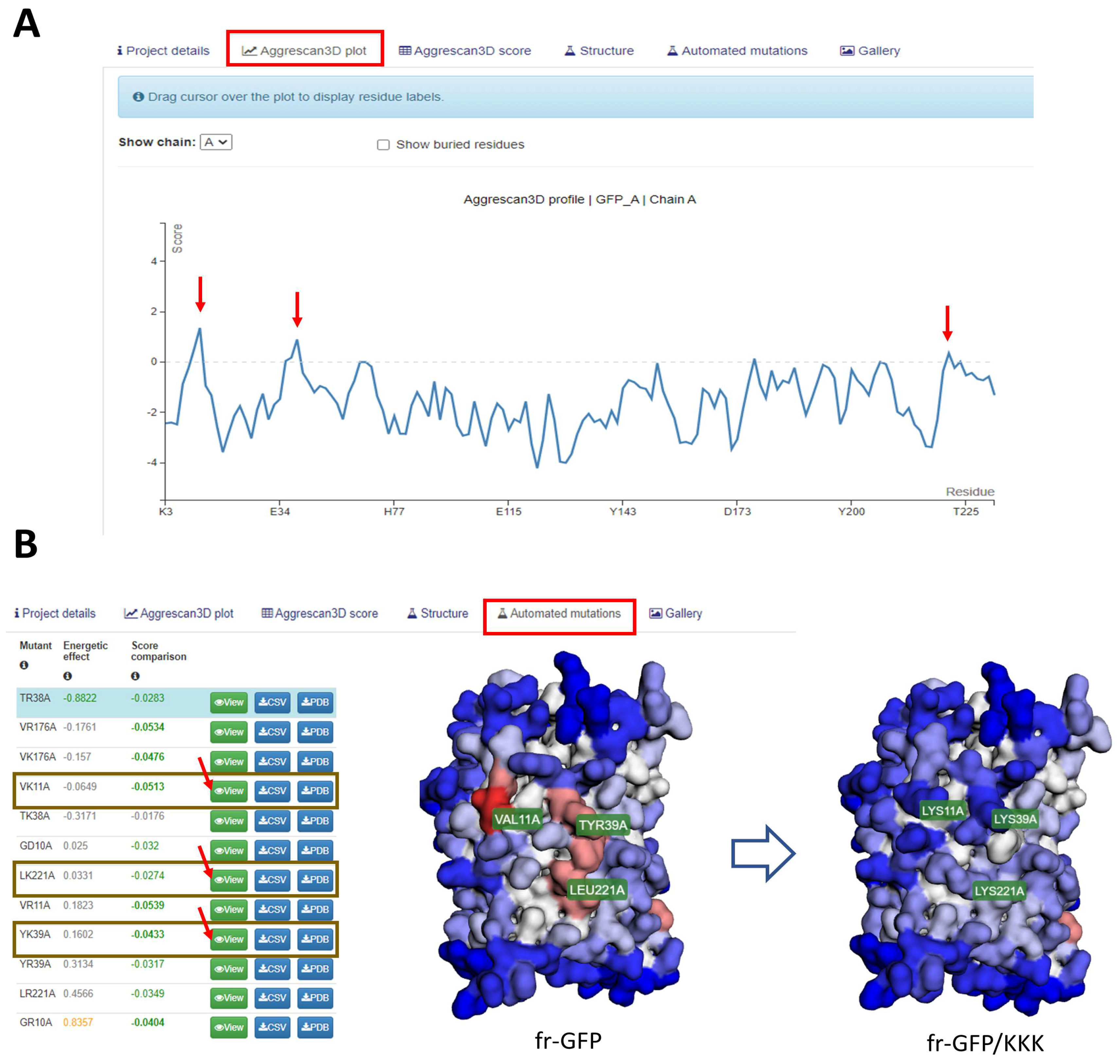
2.2. Aggrescan 3D: A Server for Prediction of Aggregation Propensity in Protein Structures and Rational Design of Protein Solubility
2.3. A3D Database: Structure-Based Predictions of Protein Aggregation for the Human Proteome
3. Computational Tools to Study Prion-like Proteins
3.1. PrionScan: An Online Database of Predicted Prion Domains in Complete Proteomes
3.2. pWaltz and PrionW: Identification of Prion-like Protein Domains
3.3. AMYCO: A Server for Prediction of the Impact of Mutations on the Aggregation Propensity of Prion-like Proteins
3.4. SGnn: A Server for the Prediction of Prion-like Domains Recruitment to Stress Granules upon Heat Stress
4. Computational Tools to Study Intrinsically Disordered Proteins (IDPs)
4.1. DispHred and DispHScan: Predicting Protein Disorder as a Function of pH
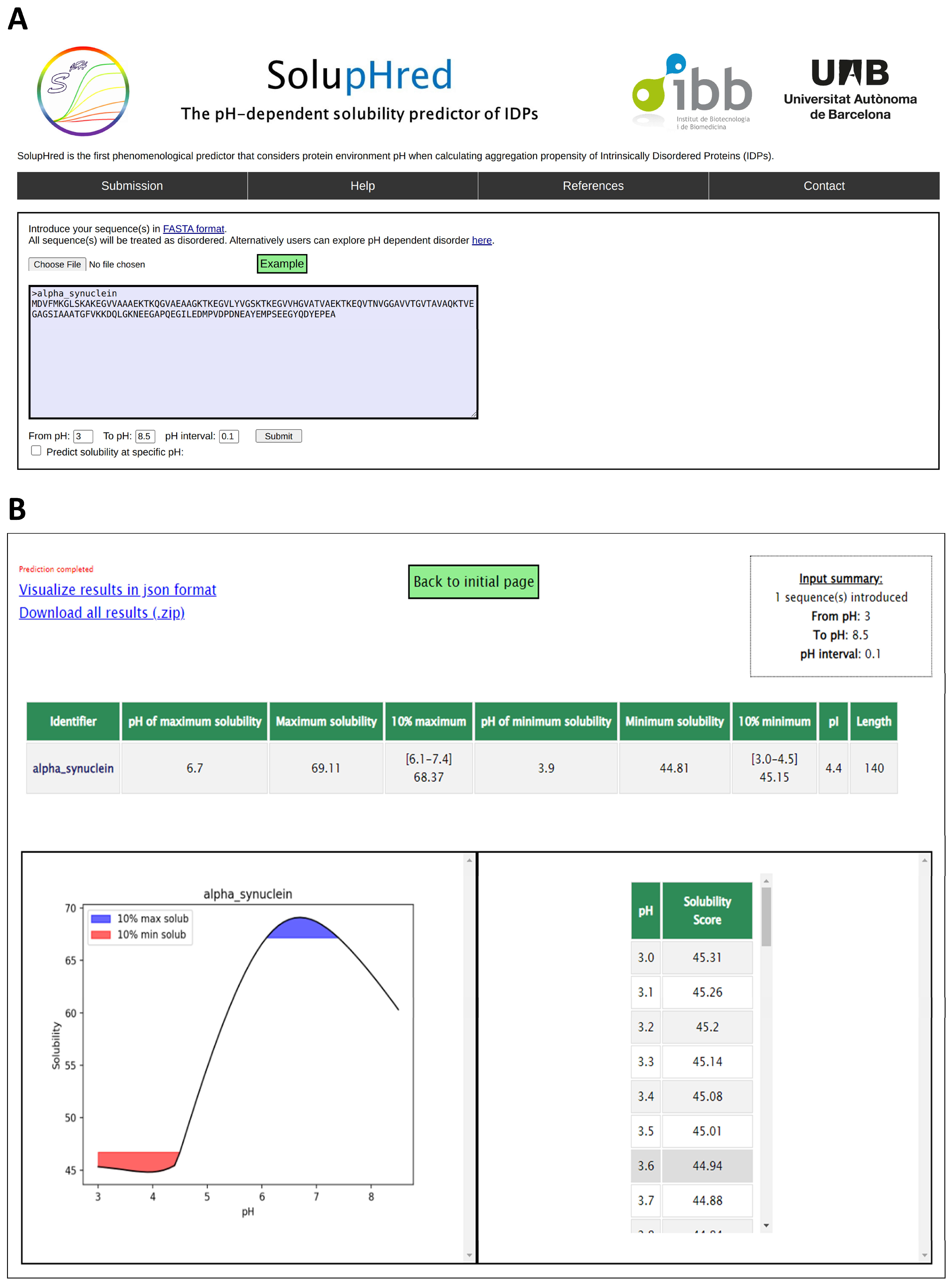
4.2. SolupHred: A Server to Predict the pH-Dependent Aggregation of Intrinsically Disordered Proteins
4.3. CARs-DB: A Database of Cryptic Amyloidogenic Regions in Intrinsically Disordered Proteins
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dill, K.A.; MacCallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daggett, V.; Fersht, A.R. Protein folding and binding: Moving into unchartered territory. Curr. Opin. Struct. Biol. 2009, 19, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Arolas, J.L.; Aviles, F.X.; Chang, J.-Y.; Ventura, S. Folding of small disulfide-rich proteins: Clarifying the puzzle. Trends Biochem. Sci. 2006, 31, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Jha, S.K. The native state conformational heterogeneity in the energy landscape of protein folding. Biophys. Chem. 2022, 283, 106761. [Google Scholar] [CrossRef]
- Pallares, I.; Vendrell, J.; Aviles, F.X.; Ventura, S. Amyloid Fibril Formation by a Partially Structured Intermediate State of α-Chymotrypsin. J. Mol. Biol. 2004, 342, 321–331. [Google Scholar] [CrossRef]
- Ventura, S.; Zurdo, J.; Narayanan, S.; Parreño, M.; Mangues, R.; Reif, B.; Chiti, F.; Giannoni, E.; Dobson, C.M.; Aviles, F.X.; et al. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc. Natl. Acad. Sci. USA 2004, 101, 7258–7263. [Google Scholar] [CrossRef] [Green Version]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Sabate, R.; Ventura, S. Cross-beta-sheet supersecondary structure in amyloid folds: Techniques for detection and characterization. Methods Mol. Biol. 2012, 932, 237–257. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. The Amyloid Phenomenon and Its Significance in Biology and Medicine. Cold Spring Harb. Perspect. Biol. 2020, 12, a033878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Fraga, H.; Graña-Montes, R.; Illa, R.; Covaleda, G.; Ventura, S. Association between Foldability and Aggregation Propensity in Small Disulfide-Rich Proteins. Antioxid. Redox Signal. 2014, 21, 368–383. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004, 342, 345–353. [Google Scholar] [CrossRef]
- Rousseau, F.; Serrano, L.; Schymkowitz, J.W. How Evolutionary Pressure against Protein Aggregation Shaped Chaperone Specificity. J. Mol. Biol. 2006, 355, 1037–1047. [Google Scholar] [CrossRef]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef]
- Ventura, S.; Villaverde, A. Protein quality in bacterial inclusion bodies. Trends Biotechnol. 2006, 24, 179–185. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Chiti, F.; Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16419–16426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baets, G.; Van Doorn, L.; Rousseau, F.; Schymkowitz, J. Increased Aggregation Is More Frequently Associated to Human Disease-Associated Mutations Than to Neutral Polymorphisms. PLoS Comput. Biol. 2015, 11, e1004374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, P.; Navarro, S.; Graña-Montes, R.; Bañó-Polo, M.; Fernández, M.R.; Papaleo, E.; Ventura, S. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biol. 2018, 14, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromwell, M.E.M.; Hilario, E.; Jacobson, F. Protein aggregation and bioprocessing. AAPS J. 2006, 8, E572–E579. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Hamrang, Z.; Rattray, N.J.; Pluen, A. Proteins behaving badly: Emerging technologies in profiling biopharmaceutical aggregation. Trends Biotechnol. 2013, 31, 448–458. [Google Scholar] [CrossRef]
- Castillo, V.; Graña-Montes, R.; Ventura, S. The aggregation properties of Escherichia coli proteins associated with their cellular abundance. Biotechnol. J. 2011, 6, 752–760. [Google Scholar] [CrossRef]
- Ciryam, P.; Tartaglia, G.G.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Widespread Aggregation and Neurodegenerative Diseases Are Associated with Supersaturated Proteins. Cell Rep. 2013, 5, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. A Relationship between mRNA Expression Levels and Protein Solubility in E. coli. J. Mol. Biol. 2009, 388, 381–389. [Google Scholar] [CrossRef]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- de Groot, N.S.; Aviles, F.X.; Vendrell, J.; Ventura, S. Mutagenesis of the central hydrophobic cluster in Abeta42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006, 273, 658–668. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Pallarés, I.; Avilés, F.X.; Vendrell, J.; Ventura, S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct. Biol. 2005, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaczewska-Dawid, A.E.; Garcia-Pardo, J.; Kuriata, A.; Pujols, J.; Ventura, S.; Kmiecik, S. A3D database: Structure-based predictions of protein aggregation for the human proteome. Bioinformatics 2022, 38, 3121–3123. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Castillo, V.; Graña-Montes, R.; Ventura, S. AGGRESCAN: Method, Application, and Perspectives for Drug Design. Methods Mol. Biol. 2012, 819, 199–220. [Google Scholar] [CrossRef]
- Tsolis, A.C.; Papandreou, N.C.; Iconomidou, V.A.; Hamodrakas, S.J. A Consensus Method for the Prediction of ‘Aggregation-Prone’ Peptides in Globular Proteins. PLoS ONE 2013, 8, e54175. [Google Scholar] [CrossRef] [Green Version]
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 103. [Google Scholar] [CrossRef]
- Martinez-Rubio, D.; Rodriguez-Prieto, A.; Sancho, P.; Navarro-Gonzalez, C.; Gorria-Redondo, N.; Miquel-Leal, J.; Marco-Marin, C.; Jenkins, A.; Soriano-Navarro, M.; Hernandez, A.; et al. Protein misfolding and clearance in the pathogenesis of a new infantile onset ataxia caused by mutations in PRDX3. Hum. Mol. Genet. 2022, 31, 3897–3913. [Google Scholar] [CrossRef]
- Tavassoly, O.; Safavi, F.; Tavassoly, I. Seeding Brain Protein Aggregation by SARS-CoV-2 as a Possible Long-Term Complication of COVID-19 Infection. ACS Chem. Neurosci. 2020, 11, 3704–3706. [Google Scholar] [CrossRef]
- De Groot, N.S.; Ventura, S. Protein Aggregation Profile of the Bacterial Cytosol. PLoS ONE 2010, 5, e9383. [Google Scholar] [CrossRef]
- Graña-Montes, R.; de Oliveira, R.S.; Ventura, S. Protein aggregation profile of the human kinome. Front. Physiol. 2012, 3, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, V.; Chiti, F.; Ventura, S. The N-terminal Helix Controls the Transition between the Soluble and Amyloid States of an FF Domain. PLoS ONE 2013, 8, e58297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Iglesias, V.; Ventura, S. Computational prediction and redesign of aberrant protein oligomerization. Prog. Mol. Biol. Transl. Sci. 2020, 169, 43–83. [Google Scholar] [CrossRef] [PubMed]
- Castillo, V.; Ventura, S. Amyloidogenic Regions and Interaction Surfaces Overlap in Globular Proteins Related to Conformational Diseases. PLoS Comput. Biol. 2009, 5, e1000476. [Google Scholar] [CrossRef] [Green Version]
- Castillo, V.; Espargaró, A.; Gordo, V.; Vendrell, J.; Ventura, S. Deciphering the role of the thermodynamic and kinetic stabilities of SH3 domains on their aggregation inside bacteria. Proteomics 2010, 10, 4172–4185. [Google Scholar] [CrossRef]
- Graña-Montes, R.; de Groot, N.S.; Castillo, V.; Sancho, J.; Velazquez-Campoy, A.; Ventura, S.; Fraga, H.; Illa, R.; Covaleda, G. Contribution of Disulfide Bonds to Stability, Folding, and Amyloid Fibril Formation: The PI3-SH3 Domain Case. Antioxid. Redox Signal. 2012, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Jamroz, M.; Kolinski, A.; Kmiecik, S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013, 41, W427–W431. [Google Scholar] [CrossRef]
- Kuriata, A.; Iglesias, V.; Pujols, J.; Kurcinski, M.; Kmiecik, S.; Ventura, S. Aggrescan3D (A3D) 2.0: Prediction and engineering of protein solubility. Nucleic Acids Res. 2019, 47, W300–W307. [Google Scholar] [CrossRef]
- Pujols, J.; Iglesias, V.; Santos, J.; Kuriata, A.; Kmiecik, S.; Ventura, S. A3D 2.0 Update for the Prediction and Optimization of Protein Solubility. Methods Mol. Biol. 2022, 2406, 65–84. [Google Scholar] [CrossRef]
- Parladé, E.; Voltà-Durán, E.; Cano-Garrido, O.; Sánchez, J.M.; Unzueta, U.; López-Laguna, H.; Serna, N.; Cano, M.; Rodríguez-Mariscal, M.; Vazquez, E.; et al. An In Silico Methodology That Facilitates Decision Making in the Engineering of Nanoscale Protein Materials. Int. J. Mol. Sci. 2022, 23, 4958. [Google Scholar] [CrossRef]
- Kuriata, A.; Iglesias, V.; Kurcinski, M.; Ventura, S.; Kmiecik, S. Aggrescan3D standalone package for structure-based prediction of protein aggregation properties. Bioinformatics 2019, 35, 3834–3835. [Google Scholar] [CrossRef]
- Gil-Garcia, M.; Baño-Polo, M.; Varejao, N.; Jamroz, M.; Kuriata, A.; Caballero, M.D.; Lascorz, J.; Morel, B.; Navarro, S.; Reverter, D.; et al. Combining Structural Aggregation Propensity and Stability Predictions to Redesign Protein Solubility. Mol. Pharm. 2018, 15, 3846–3859. [Google Scholar] [CrossRef]
- Ebo, J.S.; Saunders, J.C.; Devine, P.W.A.; Gordon, A.M.; Warwick, A.S.; Schiffrin, B.; Chin, S.E.; England, E.; Button, J.D.; Lloyd, C.; et al. An in vivo platform to select and evolve aggregation-resistant proteins. Nat. Commun. 2020, 11, 1816. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Kumru, O.S.; Blaber, S.I.; Middaugh, C.R.; Li, L.; Ornitz, D.M.; Sutherland, M.A.; Tenorio, C.A.; Blaber, M. Engineering a Cysteine-Free Form of Human Fibroblast Growth Factor-1 for “Second Generation” Therapeutic Application. J. Pharm. Sci. 2016, 105, 1444–1453. [Google Scholar] [CrossRef] [Green Version]
- Bhandare, V.V.; Ramaswamy, A. The proteinopathy of D169G and K263E mutants at the RNA Recognition Motif (RRM) domain of tar DNA-binding protein (tdp43) causing neurological disorders: A computational study. J. Biomol. Struct. Dyn. 2018, 36, 1075–1093. [Google Scholar] [CrossRef]
- Žerovnik, E. Putative alternative functions of human stefin B (cystatin B): Binding to amyloid-beta, membranes, and copper. J. Mol. Recognit. 2016, 30, e2562. [Google Scholar] [CrossRef]
- Katina, N.S.; Balobanov, V.A.; Ilyina, N.B.; Vasiliev, V.D.; Marchenkov, V.V.; Glukhov, A.S.; Nikulin, A.D.; Bychkova, V.E. sw ApoMb Amyloid Aggregation under Nondenaturing Conditions: The Role of Native Structure Stability. Biophys. J. 2017, 113, 991–1001. [Google Scholar] [CrossRef] [Green Version]
- Behbahanipour, M.; García-Pardo, J.; Ventura, S. Decoding the role of coiled-coil motifs in human prion-like proteins. Prion 2021, 15, 143–154. [Google Scholar] [CrossRef]
- Gil-Garcia, M.; Navarro, S.; Ventura, S. Coiled-coil inspired functional inclusion bodies. Microb. Cell Factories 2020, 19, 117. [Google Scholar] [CrossRef]
- Pulido, D.; Arranz-Trullén, J.; Prats-Ejarque, G.; Velázquez, D.; Torrent, M.; Moussaoui, M.; Boix, E. Insights into the Antimicrobial Mechanism of Action of Human RNase6: Structural Determinants for Bacterial Cell Agglutination and Membrane Permeation. Int. J. Mol. Sci. 2016, 17, 552. [Google Scholar] [CrossRef] [Green Version]
- Pulido, P.; Llamas, E.; Llorente, B.; Ventura, S.; Wright, L.P.; Rodríguez-Concepción, M. Specific Hsp100 Chaperones Determine the Fate of the First Enzyme of the Plastidial Isoprenoid Pathway for Either Refolding or Degradation by the Stromal Clp Protease in Arabidopsis. PLoS Genet. 2016, 12, e1005824. [Google Scholar] [CrossRef]
- Ishwarlall, T.Z.; Adeleke, V.T.; Maharaj, L.; Okpeku, M.; Adeniyi, A.A.; Adeleke, M.A. Identification of potential candidate vaccines against Mycobacterium ulcerans based on the major facilitator superfamily transporter protein. Front. Immunol. 2022, 13, 1023558. [Google Scholar] [CrossRef]
- Flores-León, M.; Lázaro, D.F.; Shvachiy, L.; Krisko, A.; Outeiro, T.F. In silico analysis of the aggregation propensity of the SARS-CoV-2 proteome: Insight into possible cellular pathologies. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140693. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Angarica, V.E.; Angulo, A.; Giner, A.; Losilla, G.; Ventura, S.; Sancho, J. PrionScan: An online database of predicted prion domains in complete proteomes. BMC Genom. 2014, 15, 102. [Google Scholar] [CrossRef] [Green Version]
- Navarro, S.; Ventura, S. Computational methods to predict protein aggregation. Curr. Opin. Struct. Biol. 2022, 73, 102343. [Google Scholar] [CrossRef]
- Iglesias, V.; de Groot, N.S.; Ventura, S. Computational analysis of candidate prion-like proteins in bacteria and their role. Front. Microbiol. 2015, 6, 1123. [Google Scholar] [CrossRef]
- Batlle, C.; Iglesias, V.; Navarro, S.; Ventura, S. Prion-like proteins and their computational identification in proteomes. Expert Rev. Proteom. 2017, 14, 335–350. [Google Scholar] [CrossRef]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.P.; DuBay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “Aggregation-prone” and “Aggregation-susceptible” Regions in Proteins Associated with Neurodegenerative Diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Angarica, V.E.; Ventura, S.; Sancho, J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genom. 2013, 14, 316. [Google Scholar] [CrossRef] [Green Version]
- O’Carroll, A.; Coyle, J.; Gambin, Y. Prions and Prion-like assemblies in neurodegeneration and immunity: The emergence of universal mechanisms across health and disease. Semin. Cell Dev. Biol. 2020, 99, 115–130. [Google Scholar] [CrossRef]
- Harbi, D.; Harrison, P.M. Interaction Networks of Prion, Prionogenic and Prion-Like Proteins in Budding Yeast, and Their Role in Gene Regulation. PLoS ONE 2014, 9, e100615. [Google Scholar] [CrossRef] [Green Version]
- Pancsa, R.; Vranken, W.; Mészáros, B. Computational resources for identifying and describing proteins driving liquid–liquid phase separation. Brief. Bioinform. 2021, 22, bbaa408. [Google Scholar] [CrossRef]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What Makes a Protein Sequence a Prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez De La Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid Fibrils of the HET-s(218–289) Prion Form a β Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef]
- Wasmer, C.; Zimmer, A.; Sabaté, R.; Soragni, A.; Saupe, S.J.; Ritter, C.; Meier, B.H. Structural Similarity between the Prion Domain of HET-s and a Homologue Can Explain Amyloid Cross-Seeding in Spite of Limited Sequence Identity. J. Mol. Biol. 2010, 402, 311–325. [Google Scholar] [CrossRef]
- Ross, E.D.; Edskes, H.K.; Terry, M.J.; Wickner, R.B. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA 2005, 102, 12825–12830. [Google Scholar] [CrossRef] [Green Version]
- Toombs, J.A.; Petri, M.; Paul, K.R.; Kan, G.Y.; Ben-Hur, A.; Ross, E.D. De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. USA 2012, 109, 6519–6524. [Google Scholar] [CrossRef] [Green Version]
- Toombs, J.A.; McCarty, B.R.; Ross, E.D. Compositional Determinants of Prion Formation in Yeast. Mol. Cell. Biol. 2010, 30, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Batlle, C.; de Groot, N.S.; Iglesias, V.; Navarro, S.; Ventura, S. Characterization of Soft Amyloid Cores in Human Prion-Like Proteins. Sci. Rep. 2017, 7, 12134. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Fernández, M.R.; Batlle, C.; Navarro, S.; de Groot, N.S.; Serpell, L.; Ventura, S. Characterization of Amyloid Cores in Prion Domains. Sci. Rep. 2016, 6, srep34274. [Google Scholar] [CrossRef]
- Pallarès, I.; Iglesias, V.; Ventura, S. The Rho Termination Factor of Clostridium botulinum Contains a Prion-Like Domain with a Highly Amyloidogenic Core. Front. Microbiol. 2016, 6, 1516. [Google Scholar] [CrossRef] [Green Version]
- Pallarès, I.; de Groot, N.S.; Iglesias, V.; Sant’Anna, R.; Biosca, A.; Fernàndez-Busquets, X.; Ventura, S. Discovering Putative Prion-Like Proteins in Plasmodium falciparum: A Computational and Experimental Analysis. Front. Microbiol. 2018, 9, 1737. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, V.; Conchillo-Sole, O.; Batlle, C.; Ventura, S. AMYCO: Evaluation of mutational impact on prion-like proteins aggregation propensity. BMC Bioinform. 2019, 20, 24. [Google Scholar] [CrossRef]
- Navarro, S.; Marinelli, P.; Diaz-Caballero, M.; Ventura, S. The prion-like RNA-processing protein HNRPDL forms inherently toxic amyloid-like inclusion bodies in bacteria. Microb. Cell Factories 2015, 14, 102. [Google Scholar] [CrossRef]
- Batlle, C.; Calvo, I.; Iglesias, V.; Lynch, C.J.; Gil-Garcia, M.; Serrano, M.; Ventura, S. MED15 prion-like domain forms a coiled-coil responsible for its amyloid conversion and propagation. Commun. Biol. 2021, 4, 414. [Google Scholar] [CrossRef]
- Zambrano, R.; Conchillo-Sole, O.; Iglesias, V.; Illa, R.; Rousseau, F.; Schymkowitz, J.; Sabate, R.; Daura, X.; Ventura, S. PrionW: A server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res. 2015, 43, W331–W337. [Google Scholar] [CrossRef] [Green Version]
- Dunn, M.J.; Shazib, S.U.A.; Simonton, E.; Slot, J.C.; Anderson, M.Z. Architectural groups of a subtelomeric gene family evolve along distinct paths in Candida albicans. G3 (Bethesda) 2022, 12, jkac283. [Google Scholar] [CrossRef]
- Du, Z.; Regan, J.; Bartom, E.; Wu, W.-S.; Zhang, L.; Goncharoff, D.K.; Li, L. Elucidating the regulatory mechanism of Swi1 prion in global transcription and stress responses. Sci. Rep. 2020, 10, 21838. [Google Scholar] [CrossRef]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef]
- An, L.; Fitzpatrick, D.; Harrison, P.M. Emergence and evolution of yeast prion and prion-like proteins. BMC Evol. Biol. 2016, 16, 24. [Google Scholar] [CrossRef] [Green Version]
- Papanikos, F.; Daniel, K.; Goercharn-Ramlal, A.; Fei, J.-F.; Kurth, T.; Wojtasz, L.; Dereli, I.; Fu, J.; Penninger, J.; Habermann, B.; et al. The enigmatic meiotic dense body and its newly discovered component, SCML1, are dispensable for fertility and gametogenesis in mice. Chromosoma 2017, 126, 399–415. [Google Scholar] [CrossRef]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Paul, K.R.; Molliex, A.; Cascarina, S.; Boncella, A.E.; Taylor, J.P.; Ross, E.D. Effects of Mutations on the Aggregation Propensity of the Human Prion-Like Protein hnRNPA2B1. Mol. Cell. Biol. 2017, 37, e00652-16. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Paul, K.R.; Hendrich, C.G.; Waechter, A.; Harman, M.R.; Ross, E.D. Generating new prions by targeted mutation or segment duplication. Proc. Natl. Acad. Sci. USA 2015, 112, 8584–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, N.M.; Naslavsky, M.; Licinio, L.; Kok, F.; Schlesinger, D.; Vainzof, M.; Sanchez, N.; Kitajima, J.P.; Gal, L.; Cavaçana, N.; et al. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G). Hum. Mol. Genet. 2014, 23, 4103–4110. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, Y.-C.; Jeong, B.-H. Novel Single Nucleotide Polymorphisms (SNPs) and Genetic Features of the Prion Protein Gene (PRNP) in Quail (Coturnix japonica). Front. Vet. Sci. 2022, 9, 870735. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-J.; Kim, Y.-C.; Kim, A.-D.; Jeong, B.-H. Novel Polymorphisms and Genetic Characteristics of the Prion Protein Gene (PRNP) in Dogs—A Resistant Animal of Prion Disease. Int. J. Mol. Sci. 2020, 21, 4160. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Won, S.-Y.; Do, K.; Jeong, B.-H. Identification of the novel polymorphisms and potential genetic features of the prion protein gene (PRNP) in horses, a prion disease-resistant animal. Sci. Rep. 2020, 10, 8926. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Kim, Y.-C.; Won, S.-Y.; Jeong, B.-H. Potential scrapie-associated polymorphisms of the prion protein gene (PRNP) in Korean native black goats. Sci. Rep. 2019, 9, 15293. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, Y.-C.; Jeong, B.-H. Novel Polymorphisms and Genetic Characteristics of the Prion Protein Gene in Pheasants. Front. Vet. Sci. 2022, 9, 935476. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Won, S.-Y.; Jeong, B.-H. Identification of Prion Disease-Related Somatic Mutations in the Prion Protein Gene (PRNP) in Cancer Patients. Cells 2020, 9, 1480. [Google Scholar] [CrossRef]
- Batlle, C.; Yang, P.; Coughlin, M.; Messing, J.; Pesarrodona, M.; Szulc, E.; Salvatella, X.; Kim, H.J.; Taylor, J.P.; Ventura, S. hnRNPDL Phase Separation Is Regulated by Alternative Splicing and Disease-Causing Mutations Accelerate Its Aggregation. Cell Rep. 2020, 30, 1117–1128.e5. [Google Scholar] [CrossRef] [Green Version]
- Boncella, A.E.; Shattuck, J.E.; Cascarina, S.M.; Paul, K.R.; Baer, M.H.; Fomicheva, A.; Lamb, A.K.; Ross, E.D. Composition-based prediction and rational manipulation of prion-like domain recruitment to stress granules. Proc. Natl. Acad. Sci. USA 2020, 117, 5826–5835. [Google Scholar] [CrossRef]
- Iglesias, V.; Santos, J.; Santos-Suárez, J.; Pintado-Grima, C.; Ventura, S. SGnn: A Web Server for the Prediction of Prion-Like Domains Recruitment to Stress Granules Upon Heat Stress. Front. Mol. Biosci. 2021, 8, 718301. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The CamSol Method of Rational Design of Protein Mutants with Enhanced Solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef]
- Luo, J.; Harrison, P.M. Evolution of sequence traits of prion-like proteins linked to amyotrophic lateral sclerosis (ALS). PeerJ 2022, 10, e14417. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Iglesias, V.; Pintado, C.; Santos-Suárez, J.; Ventura, S. DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 5814. [Google Scholar] [CrossRef]
- Zamora, W.J.; Campanera, J.M.; Luque, F.J. Development of a Structure-Based, pH-Dependent Lipophilicity Scale of Amino Acids from Continuum Solvation Calculations. J. Phys. Chem. Lett. 2019, 10, 883–889. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.-L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A.K. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15 (Suppl. 17), S4. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.; Asher, M.S.; Ehm, T.; Ionita, I.A.; Shinar, H.; Azoulay-Ginsburg, S.; Zemach, I.; Koren, G.; Danino, D.; Kozlov, M.M.; et al. Order from Disorder with Intrinsically Disordered Peptide Amphiphiles. J. Am. Chem. Soc. 2021, 143, 11879–11888. [Google Scholar] [CrossRef] [PubMed]
- Pezzotti, G.; Ohgitani, E.; Fujita, Y.; Imamura, H.; Shin-Ya, M.; Adachi, T.; Yamamoto, T.; Kanamura, N.; Marin, E.; Zhu, W.; et al. Raman Fingerprints of the SARS-CoV-2 Delta Variant and Mechanisms of Its Instantaneous Inactivation by Silicon Nitride Bioceramics. ACS Infect. Dis. 2022, 8, 1563–1581. [Google Scholar] [CrossRef] [PubMed]
- De Cena, G.L.; Scavassa, B.V.; Conceição, K. In Silico Prediction of Anti-Infective and Cell-Penetrating Peptides from Thalassophryne nattereri Natterin Toxins. Pharmaceuticals 2022, 15, 1141. [Google Scholar] [CrossRef]
- Gomari, M.M.; Rostami, N.; Faradonbeh, D.R.; Asemaneh, H.R.; Esmailnia, G.; Arab, S.; Farsimadan, M.; Hosseini, A.; Dokholyan, N.V. Evaluation of pH change effects on the HSA folding and its drug binding characteristics, a computational biology investigation. Proteins 2022, 90, 1908–1925. [Google Scholar] [CrossRef]
- Pintado-Grima, C.; Iglesias, V.; Santos, J.; Uversky, V.N.; Ventura, S. DispHScan: A Multi-Sequence Web Tool for Predicting Protein Disorder as a Function of pH. Biomolecules 2021, 11, 1596. [Google Scholar] [CrossRef] [PubMed]
- Pintado-Grima, C.; Bárcenas, O.; Ventura, S. In-Silico Analysis of pH-Dependent Liquid-Liquid Phase Separation in Intrinsically Disordered Proteins. Biomolecules 2022, 12, 974. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.M.; McGlinchey, R.P.; Lee, J.C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. USA 2010, 107, 21447–21452. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wu, Z.; Wangb, Y.; Ding, L.; Wang, Y. Role of pH-induced structural change in protein aggregation in foam fractionation of bovine serum albumin. Biotechnol. Rep. 2016, 9, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Intrinsically Disordered Proteins and Their Environment: Effects of Strong Denaturants, Temperature, pH, Counter Ions, Membranes, Binding Partners, Osmolytes, and Macromolecular Crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Pintado, C.; Santos, J.; Iglesias, V.; Ventura, S. SolupHred: A server to predict the pH-dependent aggregation of intrinsically disordered proteins. Bioinformatics 2020, 37, 1602–1603. [Google Scholar] [CrossRef]
- Santos, J.; Iglesias, V.; Santos-Suárez, J.; Mangiagalli, M.; Brocca, S.; Pallarès, I.; Ventura, S. pH-Dependent Aggregation in Intrinsically Disordered Proteins Is Determined by Charge and Lipophilicity. Cells 2020, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a Partially Folded Intermediate in α-Synuclein Fibril Formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.; Snell, J.M.; Sheftic, S.R.; Patil, S.M.; Daniels, S.B.; Kolling, F.W.; Alexandrescu, A.T. pH Dependence of Amylin Fibrillization. Biochemistry 2014, 53, 300–310. [Google Scholar] [CrossRef]
- Hortschansky, P.; Schroeckh, V.; Christopeit, T.; Zandomeneghi, G.; Fändrich, M. The aggregation kinetics of Alzheimer’s β-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 2005, 14, 1753–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; von Bergen, M.; Mandelkow, E.-M.; Mandelkow, E. The Natively Unfolded Character of Tau and Its Aggregation to Alzheimer-like Paired Helical Filaments. Biochemistry 2008, 47, 10526–10539. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Pallarès, I.; Iglesias, V.; Ventura, S. Cryptic amyloidogenic regions in intrinsically disordered proteins: Function and disease association. Comput. Struct. Biotechnol. J. 2021, 19, 4192–4206. [Google Scholar] [CrossRef]
- Pintado-Grima, C.; Bárcenas, O.; Manglano-Artuñedo, Z.; Vilaça, R.; Macedo-Ribeiro, S.; Pallarès, I.; Santos, J.; Ventura, S. CARs-DB: A Database of Cryptic Amyloidogenic Regions in Intrinsically Disordered Proteins. Front. Mol. Biosci. 2022, 9, 882160. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Caballero, M.; Navarro, S.; Fuentes, I.; Teixidor, F.; Ventura, S. Minimalist Prion-Inspired Polar Self-Assembling Peptides. ACS Nano 2018, 12, 5394–5407. [Google Scholar] [CrossRef]
- Díaz-Caballero, M.; Navarro, S.; Ventura, S. Functionalized Prion-Inspired Amyloids for Biosensor Applications. Biomacromolecules 2021, 22, 2822–2833. [Google Scholar] [CrossRef]
- Peccati, F.; Díaz-Caballero, M.; Navarro, S.; Rodríguez-Santiago, L.; Ventura, S.; Sodupe, M. Atomistic fibrillar architectures of polar prion-inspired heptapeptides. Chem. Sci. 2020, 11, 13143–13151. [Google Scholar] [CrossRef]
- Louros, N.; Orlando, G.; De Vleeschouwer, M.; Rousseau, F.; Schymkowitz, J. Structure-based machine-guided mapping of amyloid sequence space reveals uncharted sequence clusters with higher solubilities. Nat. Commun. 2020, 11, 3314. [Google Scholar] [CrossRef]
- Gondelaud, F.; Pesce, G.; Nilsson, J.F.; Bignon, C.; Ptchelkine, D.; Gerlier, D.; Mathieu, C.; Longhi, S. Functional benefit of structural disorder for the replication of measles, Nipah and Hendra viruses. Essays Biochem. 2022, 66, 915–934. [Google Scholar] [CrossRef]
- Ventura, S. Sequence determinants of protein aggregation: Tools to increase protein solubility. Microb. Cell Factories 2005, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Ninomiya, K.; Nakagawa, S.; Yamazaki, T. A guide to membraneless organelles and their various roles in gene regulation. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, F.; Santos, J.; Ventura, S. AlphaFold and the amyloid landscape. J. Mol. Biol. 2021, 433, 167059. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.A.; Craig, P.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2022, 51, D488–D508. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintado-Grima, C.; Bárcenas, O.; Bartolomé-Nafría, A.; Fornt-Suñé, M.; Iglesias, V.; Garcia-Pardo, J.; Ventura, S. A Review of Fifteen Years Developing Computational Tools to Study Protein Aggregation. Biophysica 2023, 3, 1-20. https://doi.org/10.3390/biophysica3010001
Pintado-Grima C, Bárcenas O, Bartolomé-Nafría A, Fornt-Suñé M, Iglesias V, Garcia-Pardo J, Ventura S. A Review of Fifteen Years Developing Computational Tools to Study Protein Aggregation. Biophysica. 2023; 3(1):1-20. https://doi.org/10.3390/biophysica3010001
Chicago/Turabian StylePintado-Grima, Carlos, Oriol Bárcenas, Andrea Bartolomé-Nafría, Marc Fornt-Suñé, Valentín Iglesias, Javier Garcia-Pardo, and Salvador Ventura. 2023. "A Review of Fifteen Years Developing Computational Tools to Study Protein Aggregation" Biophysica 3, no. 1: 1-20. https://doi.org/10.3390/biophysica3010001