
Amyloid-β Oligomers: Multiple Moving Targets


1
Molecular Engineering Program, University of Washington, Seattle, WA 98195, USA
2
Department of Bioengineering, University of Washington, Seattle, WA 98195, USA
*
Author to whom correspondence should be addressed.
Biophysica 2022, 2(2), 91-110; https://doi.org/10.3390/biophysica2020010
Submission received: 7 April 2022
/
Revised: 27 April 2022
/
Accepted: 27 April 2022
/
Published: 28 April 2022
(This article belongs to the Special Issue Protein Engineering: The Present and the Future)
Abstract
:Alzheimer’s Disease (AD) is a neurodegenerative disorder that is characterized clinically by progressive cognitive decline and pathologically by the β-sheet rich fibril plaque deposition of the amyloid-β (Aβ) peptide in the brain. While plaques are a hallmark of AD, plaque burden is not correlated with cognitive impairment. Instead, Aβ oligomers formed during the aggregation process represent the main agents of neurotoxicity, which occurs 10–20 years before patients begin to show symptoms. These oligomers are dynamic in nature and represented by a heterogeneous distribution of aggregates ranging from low- to high-molecular weight, some of which are toxic while others are not. A major difficulty in determining the pathological mechanism(s) of Aβ, developing reliable diagnostic markers for early-stage detection, as well as effective therapeutics for AD are the differentiation and characterization of oligomers formed throughout disease propagation based on their molecular features, effects on biological function, and relevance to disease propagation and pathology. Thus, it is critical to methodically identify the mechanisms of Aβ aggregation and toxicity, as well as describe the roles of different oligomers and aggregates in disease progression and molecular pathology. Here, we describe a variety of biophysical techniques used to isolate and characterize a range of Aβ oligomer populations, as well as discuss proposed mechanisms of toxicity and therapeutic interventions aimed at specific assemblies formed during the aggregation process. The approaches being used to map the misfolding and aggregation of Aβ are like what was done during the fundamental early studies, mapping protein folding pathways using combinations of biophysical techniques in concert with protein engineering. Such information is critical to the design and molecular engineering of future diagnostics and therapeutics for AD.
1. Introduction
Alzheimer’s Disease (AD) is a fatal neurodegenerative disorder clinically characterized by the progressive deterioration of memory and cognitive functions. It is the sixth leading cause of death in the US and the leading cause of dementia worldwide, affecting more than 50 million people [1,2,3]. The primary pathological indicators of AD are extracellular amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles of the tau protein [2]. The cascade of plaque deposition and tangle formation follows a pattern: starting in the entorhinal/perirhinal cortex, spreading through limbic structures and the hippocampus, and eventually reaching the frontal, temporal, and parietal cortex [4]. The process begins with the misfolding of the Aβ peptide, which is clipped from the amyloid precursor protein (APP) by α-, β-, and γ-secretase enzymes. In its monomeric form, Aβ is associated with a variety of biological functions [5,6,7,8,9,10], but it can misfold into an aggregation competent state, leading to a heterogeneous distribution of low- to high-molecular-weight oligomers that eventually form the characteristic amyloid plaques [11]. Importantly, disease progression is not correlated with amyloid plaque burden nor tau tangle formation, but rather with the presence of low molecular weight (LMW) soluble oligomers that act as the primary toxic agents [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32]. In fact, in the absence of fibrils, these LMW soluble oligomers induce toxicity and neuronal death, as demonstrated in mouse models of AD [21,26] and familial cases of AD that do not produce plaques [27]. Additionally, tau-mediated neuronal injury is a downstream component of AD progression preceded by Aβ accumulation and synaptic dysfunction, and Aβ oligomerization is the first known biochemical change that occurs (Figure 1) [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. Over the past decades, researchers have pursued various avenues to gain a molecular level understanding of Aβ misfolding and its relation to toxicity as the primary agent in AD pathogenesis [59,60,61,62,63,64,65,66].
Many labs have shown that LMW Aβ oligomers are more cytotoxic than protofibrillar and fibrillar structures and that they inhibit critical neuronal activities, including long-term potentiation (LTP), a classic model for synaptic plasticity and memory loss in vivo and in cell cultures [21,63,67,68]. Isolated soluble oligomer aggregates from in vitro and/or in vivo sources range in size from dimers to 24-mers, to even higher-ordered assemblies moving through the aggregation process [69,70,71,72]. However, some techniques may bias the oligomer profile, thereby making it difficult to correlate what is observed in the lab with what is present in the brain [73]. In this review we present studies on the oligomerization process—as characterized by a range of methods—to delve into the role that Aβ oligomers play in toxicity, the techniques that help to characterize different oligomer assemblies, and how this information can be used to better interpret the broad spectrum of Aβ-related data and propel AD research toward a potential treatment.
2. Aggregation and Characterization
Monomeric Aβ (4.5 kDa) is produced by the sequential proteolytic cleavage of APP (120 kDa) by β-secretase and γ-secretase in endosomes and at the plasma membrane [74]. α-Secretase also presents a pathway for formation of shorter Aβ fragments that are thought to be non-amyloidogenic [75]. Despite their common origin, the variants each possess different solubilities, stabilities, and biological and toxic properties. Clipping at the C-terminus by the γ-secretase results in variants ranging from Aβ43, Aβ42, Aβ40, Aβ38, and Aβ37 (where the number indicates the length of the fragment, ex. Aβ43 is residues 1–43), which are detected in cell culture and body fluids [3,12,15,16,17]. Further heterogeneity is exacted by diverse enzymatic processes by aminopeptidases, glutaminylcyclase, isomerases, and phosphorylation reactions, which all contribute to the sprawling list of 20 Aβ peptides that contribute distinctly to the intrinsic Aβ functions in the normal brain as well as aggregation and toxicity in the AD brain [75,76]. Of this list, Aβ40 is the most constitutively produced fragment in both healthy and AD patients, while the other Aβ peptides are continuously produced, but at lower levels.
Despite Aβ40 being the most abundant form, it is not the most pathologically relevant. Rather, Aβ42—with two additional hydrophobic residues Ile41 and Ala42 at the C-terminus—comprises the majority of plaque deposits, can seed fibril formation, and more robustly induces toxicity than its counterparts [77,78,79]. Additionally, there are distinct kinetic characteristics for the aggregation of Aβ40 and Aβ42, as well as different toxicity profiles and altered behavior when the two are co-incubated [80,81]. A common biomarker to confirm clinical AD diagnoses includes the ratio of the concentrations of Aβ42/Aβ40, which in the early stages of the disease is relatively high but then decreases as the disease progresses and more Aβ42 is deposited into the plaques. In addition to the complex interplay between Aβ variants, oligomer formation can occur through both primary and secondary nucleation, involving a pool of Aβ monomers assembling, or monomers that interact with Aβ aggregates to produce higher-order aggregates, respectively [82]. Worthy of Heraclitus’s Law of Change, this results in a wildly heterogeneous and dynamic distribution of oligomer species in a state of constant change.
Due to the dynamic nature of these Aβ aggregates, it is difficult to isolate homogeneous species for analysis and to verify that what has been generated synthetically recapitulates what is found in the body. Much like how the static structure of a protein does not fully represent its dynamic nature nor functional state(s), Aβ aggregates isolated under denaturing conditions or by using methods that necessarily break apart structured oligomers cannot accurately represent their state in the brain. Furthermore, the oligomers isolated from brain tissue may not be representative of those physiologically present, and instead they may be biased due to the isolation technique. It is likely that several of the identified species (from different techniques) may have similar characteristics, but the details of techniques used to isolate and characterize these aggregates are difficult to recapitulate and validate across labs.
A wide range of Aβ sizes and structures have been isolated and characterized using a variety of techniques, as outlined and depicted in Table 1 and Figure 1. The techniques employed can be separated into a few categories: (i) those that can be utilized throughout the aggregation process—nuclear magnetic resonance spectroscopy (NMR), circular dichroism spectroscopy (CD), size exclusion chromatography (SEC), polyacrylamide gel electrophoresis (PAGE), and microfluidic modulation spectroscopy (MMS-IR); (ii) those used in the early stages of aggregation where LMW and toxic oligomers dominate—single molecule fluorescence microscopy, ion-mobility separation-mass spectrometry (IM-MS), and A11 oligomer antibody binding; (iii) those used in the late stages of aggregation where HMW β-sheet rich oligomers dominate—small-angle X-ray scattering (SAXS) and OC oligomer antibody binding; (iv) and those used to characterize the high molecular weight (HMW), β-sheet-rich aggregates and plaques—atomic force microscopy (AFM) and electron microscopy (EM). Thus, it is important to utilize a variety of techniques to characterize Aβ aggregates and to test hypotheses garnered from those experiments in various biological models to confirm their physiological relevance. An interesting example of technique bias that may mischaracterize the aggregates present in solution is the use of SDS-PAGE for analysis of Aβ oligomerization. Bitan and coworkers [83] have shown that SDS artificially alters the aggregation state of Aβ by dissociating the aggregates. This finding was independently corroborated by Hepler and coworkers [84], who obtained the same LMW (dimer-trimer) bands for oligomer and fibril preparations of Aβ. Thus, the use of SDS-PAGE alone is not sufficient for assessing the aggregation states of Aβ. Alternative methods for analysis include: native gels, SEC, and single molecule fluorescence spectroscopy for isolation and size estimation; IM-MS (combined with molecular dynamics simulations) for insights into the assembly mechanism; AFM and EM for visualization of bulk morphological features; SAXS and cryo-EM for low resolution three-dimensional arrangements; NMR spectroscopy and x-ray crystallography for atomic-level details; and the use of conformation-specific probes for oligomer-specific recognition. Figure 2 illustrates the size ranges probed for each technique, ranging from monomeric to fibrillar assemblies.
2.1. Characterization Based on Size
The isolation and characterization of oligomeric species formed throughout aggregation is a crucial step in understanding the pathology and relevance of different aggregates in AD. Much effort has been focused on purification of samples from AD brain tissue or cultured AD-derived cells, mainly using non-denaturing PAGE and SEC [63,64,65]. It is important to utilize techniques that require minimal sample preparation and are non-denaturing, so that complex and heterogeneous samples can be isolated with relatively little bias. This represents a good starting point for subsequent, more specific characterizations that together provide the clearest possible window into the picture of molecular pathology.
SEC is a useful technique that characterizes distinct oligomer sizes based on a non-denaturing separation method. Recently, our lab reported [85] distinct oligomerization states of synthetic Aβ42, particularly hexamer and dodecamer species by SEC. These assemblies directly correlate with early aggregation events when toxicity is highest, preceding β-sheet formation, and eventually proceed to HMW species rich in β-sheet. As described below, dodecamers (and to a lesser extent hexamers) have been implicated by a variety of different studies as the primary toxic agent(s) found in patient-derived samples, and our studies have corroborated this potential role. Importantly, we showed that oligomers are stable for extended periods of time with storage on ice at various points in the aggregation process and that isolated peaks maintain the same molecular weight (MW) upon subsequent analyses with SEC [85]. This was a critical component of our in vitro studies, ensuring that the oligomers are stable assemblies that can be studied reliably from the same preparations using a variety of instruments and methods.
The dodecameric Aβ oligomer that we characterized in vitro is consistent with those previously isolated and characterized from the brains of 6-month-old transgenic mice that had begun to show symptoms of AD by Ashe and coworkers using both SEC and nondenaturing PAGE, which they refer to as Aβ*56 [63]. Adding the purified Aβ*56 directly into the brains of healthy, non-transgenic rats induces memory decline [63]. A commonly used metric for cognitive decline is the Morris water maze, which tests spatial learning and memory. This was one of the first studies to isolate and characterize a specific oligomeric species from animal brain and demonstrate a direct link between that species and toxicity in vivo. A critical component of AD research that is exploited in many studies is the fact that oligomers in the brain are stable and persist even into the late stages of disease [63,64,65,91,92]. This is contrary to the belief of many that these oligomers are transient and unstable, an idea likely from in vitro studies at much higher Aβ concentrations that drive plaque formation. Instead, the toxic oligomers are stable and can withstand processing while maintaining their structural and pathological features [20].
Another example that demonstrates the ability to isolate the stable oligomers for biophysical characterization has been described by Selkoe and coworkers, who used non-denaturing PAGE to isolate trimers from 7PA2 cells expressing the Indiana mutation of AD (affecting APP processing rather than an amino acid on Aβ, designated by APPV717F) and showing that the trimers affect LTP more strongly than other HMW aggregates [64]. Additionally, LTP decreases when dimers directly isolated from the brains of AD patients are added to the brains of healthy wild type rats [65]. This, however, poses the question of whether these LMW species are directly responsible for toxicity, or if they rapidly aggregate and form the putative species to induce toxicity. Nonetheless, Selkoe and coworkers present the argument that soluble, LMW oligomers are on-pathway aggregates that directly affect cell death and LTP [65].
To combat the difficulty of determining exact size distributions of Aβ40 oligomers, single molecule fluorescence spectroscopy was recently employed [93]. This method quantitatively infers the number of monomer subunits in an assembly by counting individual fluorophore-labeled Aβ peptides. This technique works at more physiologically relevant Aβ concentrations and is performed at the single molecule level so that it gives insights as to population heterogeneity rather than bulk characteristics. Single molecule fluorescence spectroscopy determines the heterogeneity implicit in a given sample rather than heterogeneity induced by preparation protocols, and it has the potential to flesh out individual assemblies responsible for toxicity. However, the question remains as to the influence of sample preparation, particularly for the more ‘sticky’ Aβ42 peptide, and the effect of fluorescent probes on which species are formed during aggregation and whether these protocols recapitulate what is present in vivo.
SAXS is another technique employed to monitor Aβ oligomerization [94]. SAXS has much lower spatial resolution; however, its temporal resolution is much better when combined with in-line rapid mixing. This combination gives critical information as to the early events in aggregation, and the results can give insights into the diameter, molecular weight, and polydispersity of oligomeric species [94]. Ryan and coworkers used SAXS to investigate the influence of Cu(II) on Aβ oligomerization, as Cu(II) is proposed to play a role in AD pathogenesis. Interestingly, they found that Aβ42 aggregates form ellipsoids in the presence of Cu(II), which recapitulates other widely reported annular structures, whereas Aβ40 forms protofibrillar structures [66,80,94,95]. SAXS presents a unique opportunity to monitor the early events in the aggregation of Aβ that spur the eventual fibrillization. Characterizing the populations of aggregates formed throughout disease progression is the first critical step in elucidating a mechanism for AD. The next step, then, is to ask how these discrete assemblies induce toxicity, and the structural basis for the mechanism.
2.2. Conformational Insights Provide a Basis for Toxicity
Purification and characterization of Aβ oligomers are essential in determining the specific assemblies in the heterogeneous distribution responsible for toxicity. Previously, it has been stated that unstructured oligomers are regarded as nontoxic [69,96,97], whereas structured oligomers are much more likely to be toxic [61,66,80,81,85]. Interestingly, Kayed and coworkers developed a polyclonal antibody (A11) that cross-reacts with a wide range of amyloid peptides and proteins in the oligomer state, independent of sequence and native starting structure [63,87,88,89]. This discovery implicates a common structural motif formed during oligomerization that is likely associated with the toxic state, as this antibody was able to mitigate toxicity of each of the oligomeric species. It is necessary, then, to study the structural characteristics of Aβ oligomers throughout aggregation to better understand the process and which features contribute to the toxicity mechanism. Several labs have undertaken this biophysical approach to studying AD, in a variety of different ways, as discussed below.
Our lab has recently presented evidence for the α-sheet hypothesis in Aβ42 aggregation and the structure of the toxic oligomers [85]. The α-sheet structure is a unique, nonstandard secondary structure with hydrogen bonding patterns and spectroscopic characteristics distinct from those of α-helix, β-sheet, or random coil [85,98,99,100,101,102,103,104,105]. The α-sheet structure was discovered in molecular dynamics simulations (MD) of amyloidogenic proteins as they unfolded and misfolded under amyloidogenic conditions. This unique conformation was predicted to be a key component of the toxic soluble oligomers and was verified as the dominant secondary structure formed during the lag phase of aggregation in vitro prior to formation of β-sheet structure [85,101,102,103,104]. Based on the α-sheet structure seen in the MD simulations, we designed complementary de novo, nontoxic, synthetic peptides. We used alternating l- and d-amino acids to engineer a stable form of α-sheet that does not aggregate and is recognized by the A11 antibody, consistent with the α-sheet structure being present in amyloid oligomers [85,101,102,103,104,105]. Furthermore, these de novo α-sheet peptides mitigate toxicity in cell culture, and specifically bind and neutralize soluble toxic oligomeric aggregates in both an ex vivo and in vivo transgenic AD mouse model, and in vivo AD C. elegans models [85]. In addition, the designed α-sheet peptides are being used to detect toxic oligomers in Aβ42 samples in PBS [85], spiked into plasma and CSF, and human CSF and plasma from AD patients [86]. The α-sheet hypothesis poses a new avenue for the conformation-specific detection of toxic aggregates in AD, and the use of sophisticated techniques to determine the conformation of aggregates may help shed light on this and other proposed mechanisms [105]. Notably, the discovery of α-sheet structure is the direct result of years of working out methods to simulate protein unfolding/folding in combination with validation via protein engineering experiments [106,107].
Regarding the conformational characterization of aggregates, IM-MS is a promising technique for interrogating the structure of oligomeric assemblies, and it has been used to suggest that Aβ40 and Aβ42 form distinct conformations [80,81]. For example, Aβ40 tetramers form a compact ring-shaped structure, making it more difficult to form further contacts. Alternatively, Aβ42 tetramers prefer a bent structure with subunits at either end that easily facilitate the addition of subsequent species. This technique showed that Aβ40 oligomers remain smaller, while the Aβ42 tetramers further assemble into large donut-shaped dodecamers, which could seed and accelerate protofibril formation and elongation [80,81]. As the dodecamers (Aβ*56) have been shown to specifically cause memory deficits [63], these IM-MS studies shed light on the higher order structure behind the biologically relevant Aβ42 toxic isoform.
AFM and EM are also useful techniques to gain insights as to the structural topology of specific aggregates. These two techniques have been used to understand the morphologies of oligomers and fibrils that form on the micrometer or nanometer scale [61,66]. AFM in particular has been used to probe the morphology of oligomers at lipid bilayers. One study demonstrated that Aβ42 oligomers assemble into circular aggregates on the order of 8–12 nm that interact with the lipid bilayers to form a membrane-permeable pore [66]. This mechanism of toxicity is specifically implicated to trigger flow through the cell membrane leading to cell death and provides an explanation for the toxic nature of the more structured Aβ oligomers over the amorphous aggregates [66,108].
2.3. Atomic Details of Toxic and Nontoxic Oligomer Conformations
The intrinsic heterogeneity and dynamic nature of Aβ oligomers have made high-resolution structural analysis of Aβ oligomers very difficult. Despite this, 2D NMR spectroscopy [22,109] and X-ray crystallography [110] have been used to probe the atomic details of kinetically trapped oligomers. Ishii and coworkers [109] proposed that despite the size heterogeneity of prefibrillar Aβ40 oligomers, they maintain parallel β-strand structure similar to amyloid fibrils. Incubating the solution at 4 °C and flash freezing them in liquid nitrogen allowed them to quickly collect 2D solid state NMR spectra at 15 °C [109]. Though the oligomers produce less overall Nuclear Overhauser Effect crosspeaks (NOEs) and form less contacts than the fibrils, they claim that the similar spectral characteristics and connectivities displayed by key amino acids throughout the peptide indicate that the underlying structures must be related despite major morphological differences. Importantly, they focus on 650 kDa, non-A11 reactive spherical aggregates that form just prior to fibril deposition, indicating that they are likely the late stage β-sheet protofibrils that seed fibril formation and are not directly associated with the physiological toxicity mechanism.
Some labs have turned to the use of non-physiological Aβ peptide fragments and protein engineering techniques to explore potential sequence-mediated aggregation mechanisms. Pham and coworkers utilized X-ray crystallography to determine a crystal structure of oligomers formed by a core Aβ segment (containing residues 15–23) and demonstrate how this region generates a wide array of oligomer assemblies, possibly due in part to the missing residues [110]. Once the discovery of self-propagating molecular-level polymorphism in Aβ fibrils was established, protocols for preparing relatively homogenous samples were developed [90,111] and structural models for Aβ40 polymorphs were produced from solid state NMR data. Smith and coworkers [22] also used solution-based NMR to acquire a number of NOEs to aid in the development of pentamer and fibril models at low temperature and low salt conditions. Unfortunately, however, atomic-level details of full-length LMW toxic Aβ oligomers under physiological conditions have yet to be elucidated due to the lack of a sufficient number of experimental observables for generation of atomic models.
2.4. Toward Characterization of Brain-Derived Oligomers
Several labs have shown that brain-derived soluble Aβ species can be extracted from AD patients using saline buffers without detergents [16,112]. The extracted samples can then be fractionated with nondenaturing SEC to isolate individual assemblies for discrete characterizations [16,112]. Using such methods, O’Nuallain and coworkers demonstrated that synaptotoxicity correlates with soluble Aβ oligomers ranging from dimers-hexamers [16,112]. Shankar and coworkers used SDS-PAGE to characterize dimeric Aβ present in AD tissue and suggest that this dimer is the minimal toxic component in vivo, but due to recognized artifacts with this technique, questions remain as to whether the toxicity is directly caused by these dimers [65]. Dissociation into low MW species by using SDS-PAGE and rapid aggregation of the putative dimers into larger aggregates might provide an alternate explanation. In a later study, these authors found that synthetically prepared dimers—used as a model for the brain-derived species—rapidly aggregate into metastable protofibrils, which suggests a much more complex process is likely at play [113]. Larger Aβ oligomers, >100 kDa and A11-negative, with spherical structures roughly 10 nm across have also been isolated from AD-affected brain tissue and been proposed as the likely toxic agents, further confounding the study of brain-derived oligomers [114,115]. These assertions are in opposition to Ashe and coworkers who have reproducibly isolated the Aβ*56 oligomer from AD brains, indicating that the LMW oligomers responsible for synaptotoxicity are present, despite not being a primary component in the pathological characterizations (which rely on plaque burden) [63]. Furthermore, Ashe and coworkers found that the larger MW oligomers are OC-positive (OC is an antibody that specifically binds the fibril plaques, Figure 1 and Figure 2) and are not toxic. Nonetheless, that this range of soluble oligomers is stable enough to be isolated from diseased human brains is again critical to the study of AD, indicating that oligomers are persistent in both the beginning and late stages of disease where the primary pathology is plaque burden.
3. Mechanisms of Toxicity by Oligomers
In recent years, the view that the low molecular weight, soluble oligomers are the primary toxic agents has become more widely accepted. Furthermore, neuronal damage begins to occur 10–20 years before presentation of the symptoms in AD patients, and amyloid plaques are late-stage pathological indicators of the disease [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66]. Due to the heterogeneous and dynamic nature of the Aβ oligomers, the mechanisms by which these aggregates carry out their toxic effects and initiate neuronal death is difficult to determine; however, increasing information about the mechanism of action of these oligomers is becoming available (Figure 3).
3.1. Membrane Interactions Foster Aβ Toxicity
It has been proposed that Aβ oligomers affect neuronal membranes by a number of different mechanisms. Synapse loss is an indication of AD and greatly contributes to the cognitive deterioration of patients, which is often preceded by attenuation of LTP, lowered synaptic plasticity, and decline of spine density [116]. These neurological features have been known for years, but recent studies have shed light on the mechanistic details.
Some studies have shown that Aβ oligomers bind to the neuronal membrane, causing local perturbations that damage membrane integrity [108,117,118]. Specifically, SDS-PAGE-isolated Aβ dimers accumulate at lipid rafts in the brains of transgenic Tg2576 mice, which overexpress the Swedish mutation (APPKM670/671NL) causing increased production of Aβ40 and Aβ42 [118]. Importantly, the dense localization of Aβ oligomers at lipid rafts increases the local concentration of Aβ and might help to seed aggregation and plaque formation, which is not found in most cases at physiologically relevant concentrations. This may help to explain why the Aβ monomers show little inclination to aggregate into the toxic oligomers at such low concentrations in healthy brains, but under certain circumstances that promote localization they misfold, aggregate, and form the toxic species. Furthermore, Selkoe and coworkers confirmed the observation that SDS-dimers interact with lipid membranes much more rapidly than their monomeric counterparts [118]. Upon immunoprecipitation, the dimers associate with GM1-gangliosides in the lipid rafts. These sites are hubs for signal transduction, and the density of oligomers in these regions necessitates the oligomer effect on signal propagation, thus aiding in memory decline. This result is further supported by the correlation of Aβ dimer localization at lipid rafts in 6-month-old Tg2576 mice with memory impairment symptoms [117]. Disregarding the assertion of dimer-specific influence here due to the issues associated with SDS-isolation, the fact that the misfolded and aggregated forms of Aβ—and not the monomeric form—localize at neuronal membranes indicates that misfolding and localization play important roles in the pathogenesis in AD.
Some studies show a more active form of toxicity exacted by Aβ oligomers at the membrane [119]. The formation of annular structures ranging from 40–170 kDa (from combined SDS-PAGE and AFM studies) insert themselves at the neuronal membrane to initiate dysregulation of efflux/influx through the creation of membrane pores [108,119,120]. The perforation of neuronal membranes has been attributed to the antiparallel β-sheet conformation of late-stage oligomers and fibrils, which disrupt cellular systems [120]. This penetration leaves large channels where passive diffusion of small molecules takes place, thus dysregulating the intracellular and extracellular environment. Alternatively, some researchers propose that the pore formation is instead due to the formation of α-helical structures linked to a repeat motif of GxxxG in the Aβ peptide [121,122]. It is proposed that this motif may facilitate the formation of α-helical structures that form pores at the membrane and disrupt molecular flux. Along these lines, as support for the possibility that the pores may contain α-sheet, we note that the potassium channel contains four α-strands with the aligned main chain carbonyl oxygens pointing toward the center of the channel to facilitate ion flow [100,123].
3.2. Intracellular Effects of Aβ Oligomers
Receptor-mediated toxicity may present an even more guided mechanism of Aβ oligomer toxicity through endocytosis at specific sites on the membrane. Several studies have demonstrated the binding of Aβ oligomers to receptors on the neuronal membrane, leading to endocytosis of oligomers into the intracellular space and initiating damage that affects neuronal signaling [124,125,126]. Treatment of astrocytes with oligomeric aggregates of Aβ specifically increases glutamate release, as detected with a FRET-based glutamate sensor; however, this effect is not observed following treatment with Aβ monomers [124]. This glutamate response is calcium-dependent and initiated by the binding of Aβ trimers (isolated by SEC) to the α7 nicotinic acetylcholine receptors of astrocytes, which is a calcium permeable ion channel [124]. The effect on synapse signaling is due largely to the overactivation of extrasynaptic N-methyl-D-aspartate receptors (eNMDAR) on hippocampal neurons following the glutamate increase. Additionally, these studies show that Aβ oligomer-receptor complexes promote internalization through endocytosis that triggers damage specifically to the intracellular compartments [126]. This complexation affects cellular signaling; for example, the activation of eNMDARs by SEC-isolated trimers increases calcium flux, which triggers nitric oxide synthase to increase NO production, which then leads to apoptosis [124]. Increased NO levels also affect synaptic spine density, where a loss often results in degeneration of neuronal synapses and thus connectivity. These complexes activate specific kinases that decrease NMDAR density and cause dendritic spine loss. In this case, Aβ oligomer toxicity is carried out by both receptor- and spine density-mediated mechanisms, purportedly due to the misfolded prion-like activity of the oligomer species [124,126].
Extracellular Aβ can interact with membranes and receptors, but it can also be internalized into neurons by endocytosis. Additionally, Aβ accumulates intracellularly where APP is present in the membrane [127,128,129]. The mitochondria, endoplasmic reticulum (ER), trans-Golgi network, endosomes, autophagosomes, and lysosomes are all implicated as potential sites for Aβ generation and aggregation. Intracellular Aβ oligomers can enact cellular damage and initiate cell death through elevated ER stress, mitochondrial damage, calcium ion dysregulation, and apoptosis [127,128,129].
Umeda and coworkers used an AβE22Δ (APPE693Δ) transgenic mouse line to express mutant Aβ (termed the Osaka mutant) that forms soluble oligomers but does not form fibrils [27,31,32,127]. They demonstrated that aggregates (of an undetermined size) colocalize at cellular organelles in neurons, and these mice have elevated levels of markers indicative of heightened ER stress [127]. This stress is specifically due to the intracellular accumulation of Aβ oligomers and activated by phospholipase C signaling, which initiates release of calcium from the ER into the cytosol [128]. Cell viability specifically decreases and caspase 3 (an apoptotic initiator) is activated in response to this dysregulation [127,129]. The Osaka mutant is a very unique case of AD, but importantly portrays the essential role that soluble oligomers play in the toxicity mechanism, even in the absence of amyloid fibrils.
This brief summary of the vast number of alternative pathways of Aβ oligomer-induced toxicity demonstrates that no single mechanism explains all aspects of toxicity in AD. The process is multifaceted and may involve a myriad of interconnected processes and signaling pathways to initiate the neurodegenerative decline characteristic of AD. Importantly, however, there is recent evidence from several groups for a more generic type of toxicity. Wogulis et al. have shown that the ongoing self-assembly of Aβ42 into fibrils from soluble monomers and oligomers is, by itself, sufficient to cause cell impairment and death [130]. Additionally, synthetic α-sheet peptides designed to specifically target the toxic species neutralize the toxicity and prevent further aggregation [85]. Thus, the dynamic aggregation-based model of toxicity is compatible with most observations in the literature; however, an exact mechanism has yet to be elucidated, but a more nuanced mechanism involving lipids facilitating membrane disruption is intriguing [131].
4. Aβ-Based AD Treatments in Clinical Trials
There have been a wide range of small molecules and antibodies that have entered clinical trials in recent decades for the treatment of Alzheimer’s disease. Trials include compounds from large companies such as Biogen, Roche, Genentech, and many more, and despite the abundance of funding and manpower dedicated to drug development, there has been little clinical success [132]. The clinical trials have taken three main approaches: (1) disrupting the fibrillar form of Aβ by binding to and breaking up plaques; (2) sequestering the monomeric form of Aβ through sequence-specific binding; or (3) halting the production of Aβ by inhibiting the β-secretase enzyme that clips APP to give rise to Aβ (BACE inhibitors) [132]. While there has been much discussion of enrolling patients earlier to obtain better outcomes, it would be inherently difficult to obtain significant clinical efficacy with any of these approaches. Targeting fibrils is not only an intervention that takes place too late in the disease, but it has also been shown to increase the level of soluble oligomers in the brain, thereby increasing toxicity rather than halting disease progression [82,132]. Similarly, targeting the monomer directly through sequence recognition or with BACE inhibitors would remove the physiologically necessary form of Aβ from the brain, thereby disrupting processes that promote normal function, not to mention affecting the processing of other peptides [5,6,7,8,9,10,132]. Recently several companies have claimed that their compounds target the soluble oligomers formed in the early stages of disease, but further analysis of the antibodies and small molecules used indicates that they are targeting late-stage β-sheet rich protofibrils, fibril plaque deposits, or they indiscriminately bind different Aβ conformers. We outline a few of the Aβ targeting approaches below.
Aducanumab™ is a monoclonal antibody that preferentially binds to the protofibrils and plaques formed in the late stages of disease (Figure 3) [132,133,134]. Aducanumab was developed using an immunotherapeutic approach wherein human B-cell clones were exposed to aggregated Aβ and screened for reactivity [133]. The resultant library went through molecular cloning, sequencing, and recombinant expression and eventually resulted in the monoclonal antibody Aducanumab, which selectively reacts with both soluble oligomers and insoluble fibrils [133]. The cross-reactivity of the monoclonal antibody for soluble and insoluble forms of aggregated Aβ indicates that the recognition motif is likely the β-sheet structure shared by late-stage protofibrillar HMW aggregates and the fibril plaque deposits. Due to the binding preference of this antibody, measures of efficacy were necessarily contingent upon initial plaque presence in a variety of animal and human cohorts and was verified using amyloid PET imaging (which uses a plaque-specific radiotracer to measure amyloid burden). Thus, enrolled patients were likely in the mid-to-late stages of the disease when they were undergoing treatment. The hope was that Aducanumab would be an effective intervention in the early stages of the disease [134], but it appears to be targeting a late-stage indicator of disease progression [132,133]. Aducanumab (marketed as Aduhelm) has recently been approved for use by the FDA but is undergoing continued scrutiny due to a lack of consistent data in clinical trials.
Solanezumab™ is a monomer-specific monoclonal antibody probe that has been discussed as a potential early-stage therapeutic option for AD patients in the mild cognitive impairment (MCI) stage (Figure 3) [132,135,136]. The parent monoclonal antibody for Solanezumab (m266) was generated in A/J mice using a synthetic Aβ13-28 peptide (HHQKLVFFAEDVGSNGGC) where several positive clones were identified that all selectively bound to Aβ1-28, but not Aβ1-16 nor Aβ17-28, suggesting specificity for residues 13-28 of Aβ [135]. Early characterization showed that this antibody was unable to bind to Aβ aggregates, but completely prevented Aβ42 fibrillization in vitro by binding and sequestering the monomeric peptide [135]. Thus, Solanezumab is considered the AD paradigm of a sequence-based antibody that selectively binds the free, not solely the aggregated, form of Aβ. Based on past clinical trials and the scientific literature surrounding Aβ functions and toxicity mechanisms, sequence-based interventions for AD will not halt nor prevent disease progression [132]. In fact, monomer-targeting compounds have been associated with various side effects, likely due to removal of the physiologically active form of the peptide from the brain [132]. This may produce positive results in the short-term, which is generally measured in animal models and early trials; however, the long-term rehabilitative effects are much more difficult to determine.
Crenezumab™ is a monoclonal antibody that has been used in several AD clinical trials, promoted as a truly oligomer-specific marker that binds to the LMW, soluble oligomers formed in the early stages of AD (Figure 3) [132,137,138,139]. Crenezumab was generated by immunizing mice with an Aβ1-15 peptide antigen and screening for antibodies that bound several different species of Aβ (including monomer) and inhibited Aβ42 assembly into small cytotoxic aggregates [139]. Early clinical studies indicated that Crenezumab specifically reduces Aβ oligomers present in CSF after treatment in humans and a variety of different animals [137,138,139]. Interestingly, however, the antibody specifically binds to protofibrillar Aβ and mature amyloid plaques, indicating that the conformational specificity is for the β-sheet structure formed in the late stages of disease [138].
Finally, Lanabecestat™ is one of many examples of BACE inhibitor therapeutics tested for AD treatment (Figure 3) [132,140]. Lanabecestat works by inhibiting the β-secretase enzyme from clipping at the β-site of APP, thus mitigating the production of Aβ monomers in the brain. Many labs have shown the efficacy of this approach in vitro, animal, and early clinical models [132,140]. Removal of Aβ prevents formation of the toxic oligomers, protofibrils, and plaques. The Lanabecestat clinical trial was eventually stopped under futility analysis, unlike similar BACE inhibitors Verubecestat™ and Atabecestat™, which were halted due to side effects [131]. Even though Lanabecestat was well tolerated, neither of the two doses tested was effective at shifting the primary or secondary outcomes from placebo [132]. Targeting BACE is an enticing strategy for treating AD, as mitigating the production of more Aβ monomers can halt further oligomer/fibril formation; however, as most patients will be treated only after showing symptoms, oligomers and fibrils are largely already present. Moreover, there are concerns about the removal of Aβ monomers given their many biological functions [5,6,7,8,9,10], as well as other peptides processed by the inhibitor secretases [3,12,15,16,17,74,75,132,140].
5. Conclusions and Outlook
AD is an amyloid disease characterized by the aggregation of the Aβ peptide monomer into a heterogeneous distribution of soluble, toxic oligomers, followed by relatively nontoxic protofibrils, and eventually results in the characteristic nontoxic cross-β pleated sheet fibrils. The toxic intermediates are dynamic in nature, making the study of their size, structure, and the detailed mechanism of toxicity difficult to characterize. The fibrils are nontoxic, but they can act as a reservoir of soluble oligomers by its fragmentation and secondary nucleation. Recent advances in biophysical and biochemical methods have provided more insights into the structural characteristics of the oligomers formed throughout aggregation. Isolation and size estimation using PAGE and SEC have led to informed studies of the effects that different size aggregates have on neuronal function. However, it must be noted that while the isolation of oligomers helps us to characterize the process, the effect of isolation is not fully known, and, in particular, as discussed above, there are known artifacts with PAGE methods. Assembly pathways have been investigated with the aid of IM-MS experiments and MD simulations. Bulk features of morphology and topology have been directly visualized with techniques like AFM and EM, while more detailed information has been obtained with NMR and X-ray crystallography. Importantly, conformation-specific probes have been developed to investigate and interfere with specifically assembled structures that are associated with toxicity.
Along with advances in oligomer characterization, many toxicity mechanisms governed by Aβ oligomers have been proposed and investigated. These mechanisms range from extracellular interactions with membranes or receptors, intracellular accumulation disrupting normal function, and cell-to-cell transmission of toxic aggregates. Researchers are shifting their focus now to the relationship between oligomer conformations and toxicity and using this information to design probes that can intervene in the early stages of disease progression for diagnosis and treatment. In this regard, it is critical for researchers to carefully report the details of their oligomer preparation protocols and experimental conditions to compare what is done in different labs to help facilitate progress in the field and ensure reproducibility.
The unfortunate failures of past AD clinical trials have been taken as a failure of the amyloid cascade hypothesis, and this in turn has spurred efforts focused on other targets. While expanding the scope to investigate other targets is overall beneficial for the field, we and others argue that it is not a failure of the amyloid cascade hypothesis per se. Instead, these failures illustrate the importance of clearly distinguishing and targeting specific Aβ conformational species, rather than collectively lumping together states that are conformationally and physically distinct into a catch-all amyloid state. To avoid confusion, it is best to reserve the term amyloid for true amyloid fibrils and plaques. Given these and other factors that have come to light in the last years [141,142], the amyloid hypothesis is better denoted as the oligomer cascade hypothesis. This highlights the importance of the early-stage toxic oligomeric species as well as the dire need for a shift in AD diagnosis and treatment toward an oligomer-based approach. The studies above have outlined the latency for AD symptoms years after neuronal damage has taken place. An associated component in the refinement of therapeutic efforts is that AD diagnostics must detect individuals at risk earlier, prior to the development of the current late-stage disease markers. In this case, new therapeutics could then be tested for their ability to mitigate disease progression, rather than trying to reverse neuronal damage that has taken place over 10–20 years. On a more sobering note, AD is incredibly complex and when it appears that we have a handle on the many effects of toxic oligomers, taking a narrow example, and then new neuron-specific deleterious activities are discovered and, as ever, the devil is in the details [143].
Author Contributions
D.S. and V.D. wrote this review. All authors have read and agreed to the published version of the manuscript.
Funding
NIH AG067476, NIH/NIBIB 532EB1650.
Acknowledgments
We are grateful for financial support from the NIH National Institute of Aging (R01 AG067476 to V.D.) and the National Institute of Health Bioengineering and Cardiovascular Training Grant (NIH/NIBIB 532EB1650 to D. S., M.Regnier P.I.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Abbreviation | Term |
| AD | Alzheimer’s Disease |
| Ab | Amyloid beta peptide |
| APP | Amyloid Precursor Protein |
| AFM | Atomic Force Microscopy |
| BACE Inhibitor | Beta-secretase inhibitor |
| CD | Circular Dichroism Spectroscopy |
| EM | Electron Microscopy |
| ER | Endoplasmic Reticulum |
| eNMDAR | Extrasynaptic N-methyl-D-aspartate Receptors |
| FTIR | Fourier Transform IR |
| Forster Resonance energy transfer | FRET |
| HMW | High Molecular Weight |
| IM-MS | Ion-Mobility Separation-Mass Spectrometry |
| LTP | Long Term Potentiation |
| LMW | Low Molecular Weight |
| MMS-IR | Microfluidic Modulation Spectroscopy |
| MCI | Mild Cognitive Impairment |
| MD | Molecular Dynamics |
| NMR | Nuclear Magentic Resonance Spectroscopy |
| NOEs | Nuclear Overhauser Effect Cross Peaks |
| PAGE | Polyacrylamide Gel Electrophoresis |
| PET | Positron Emission Tomography |
| SEC | Size Exclusion Chromatography |
| SAXS | Small-angle X-ray Scattering |
| SDS-PAGE | Sodium Dodecyl Sulfate Polycacrylamide Gel Electrophoresis |
References
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. 2022. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 27 April 2022).
- Masters, C.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; De Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of β-amyloid: Where the Alzheimer’s disease protein meets physiology. Rev. Neurosci. 2010, 21, 83–93. [Google Scholar] [CrossRef]
- Whitson, J.S.; Selkoe, D.J.; Cotman, C.W. Amyloid β Protein Enhances the Survival of Hippocampal Neurons in Vitro. Science 1989, 243, 1488–1490. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Banks, W.A.; Johnson, S.N.; Yamada, K.A.; Xu, L. A physiological role for amyloid-β protein: Enhancement of learning and memory. J. Alzheimer’s Dis. 2010, 19, 441–449. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. Physiological roles of amyloid-β and implications for its removal in Alzheimer’s disease. Drugs Aging 2004, 21, 621–630. [Google Scholar] [CrossRef]
- Hiltunen, M.; van Groen, T.; Jolkkonen, J. Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: Evidence from experimental studies. J. Alz. Dis. 2009, 18, 401–412. [Google Scholar]
- Koudinov, A.R.; Berezov, T.T. Alzheimer’s amyloid-β (Aβ) is an essential synaptic protein, not neurotoxic junk. Acta Neurobiol. Exp. 2004, 64, 71–79. [Google Scholar]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Lambert, M.P.; Viola, K.L.; Chromy, B.A.; Chang, L.; Morgan, T.E.; Yu, J.; Venton, D.L.; Krafft, G.A.; Finch, C.E.; Klein, W.L. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 2001, 79, 595–605. [Google Scholar] [CrossRef]
- Wang, J.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 1999, 158, 328–337. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2016, 37, 152–163. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef]
- Zahs, K.R.; Ashe, K.H. β-Amyloid oligomers in aging and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Jack Jr, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Neurology 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Ashe, K.H. The biogenesis and biology of amyloid β-oligomers in the brain. Alz. Dement. 2020, 16, 1561–1567. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Abeta (1–42) in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L. Synaptotoxic amyloid-β oligomers: A molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J. Alz. Dis. 2013, 33, S49–S65. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Heurling, K.; Ashton, N.J.; Scholl, M.; Zimmer, E.R. In vivo detection of Alzheimer’s disease. Yale J. Biol. Med. 2018, 91, 291–300. [Google Scholar] [PubMed]
- Counts, S.E.; Ikonomovic, M.D.; Mercado, N.; Vega, I.E.; Mufson, E.J. Biomarkers for the early detection and progression of Alzheimer’s Disease. Neurotherapeutics 2017, 14, 35–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Herukka, S.K.; Simonsen, A.H.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S.; et al. Recommendations for cerebrospinal fluid Alzheimer’s disease biomarkers in the diagnostic evaluation of mild cognitive impairment. Alz. Dem. 2017, 13, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Diniz, B.S.; Pinto Junior, J.A.; Forlenza, O.V. Do CSF total tau, phosphorylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer’s disease? A systematic review and meta-analysis of the literature. World J. Biol. Psychiatry 2008, 9, 172–182. [Google Scholar] [CrossRef]
- Ferreira, L.; Santos-Galduroz, R.F.; Ferri, C.P.; Fernandes Galduroz, J.C. Rate of cognitive decline in relation to sex after 60 years-of-age: A systematic review. Geratr. Gerontol. Int. 2014, 14, 23–31. [Google Scholar] [CrossRef]
- Ferreira, D.; Rivero-Santana, A.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarría, A.; Serrano-Aguilar, P. Improving CSF biomarkers’ performance for predicting progression from mild cognitive impairment to Alzheimer’s disease by considering different confounding factors: A metaanalysis. Front. Aging Neurosci. 2014, 6, 287. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, I.A.; Vos, S.; Handels, R.; Visser, P.J. Biomarkers as predictors for conversion from mild cognitive impairment to Alzheimer-type dementia: Implications for trial design. J. Alz. Dis. 2010, 20, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Takwoingi, Y.; Flicker, L.; Mason, S.E.; McShane, R. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 6, 8782. [Google Scholar] [CrossRef] [PubMed]
- Noel-Storr, A.H.; Flicker, L.; Ritchie, C.W.; Nguyen, G.H.; Gupta, T.; Wood, P.; Walton, J.; Desai, M.; Solomon, D.F.; Molena, E.; et al. Systematic review of the body of evidence for the use of biomarkers in the diagnosis of dementia. Alz. Dement. 2013, 9, e96–e105. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Su, B.; Zheng, H.; Kim, J.R. A peptide probe for detection of various beta-amyloid oligomers. Mol. Biosyst. 2012, 8, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhong, Y.; Gui, J.; Wang, X.; Zhuang, X.; Weng, J. A hydrogel biosensor for high selective and sensitive detection of amyloid-beta oligomers. Int. J. Nanomed. 2018, 13, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Laske, C.; Sohrabi, H.R.; Frost, S.M.; López-de-Ipiña, K.; Garrard, P.; Buscema, M.; Dauwels, J.; Soekadar, S.R.; Mueller, S.; Linnemann, C.; et al. Innovative diagnostic tools for early detection of Alzheimer’s disease. Alz. Dement. 2015, 11, 561–578. [Google Scholar] [CrossRef]
- More, S.S.; Beach, J.M.; McClelland, C.; Mokhtarzadeh, A.; Vince, R. In vivo assessment of retinal biomarkers by hyperspectral imaging: Early detection of Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 4492–4501. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Radanovic, M.; Talib, L.L.; Aprahamian, I.; Diniz, B.S.; Zetterberg, H.; Gattaz, W.F. Cerebrospinal fluid biomarkers in Alzheimer’s disease: Diagnostic accuracy and prediction of dementia. Alz. Dem. Diagn. Assess. Dis. Monit. 2015, 1, 455–463. [Google Scholar] [CrossRef]
- Nabers, A.; Hafermann, H.; Wiltfang, J.; Gerwert, K. Abeta and tau structure-based biomarkers for a blood- and CSF-based two-step recruitment strategy to identify patients with dementia due to Alzheimer’s disease. Alz. Dem. Diagn. Assess. Dis. Monit. 2019, 11, 257–263. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. J. Am. Med. Assoc. 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphortylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, 861. [Google Scholar] [CrossRef]
- Toledo, J.B.; Xia, S.X.; Trojanowski, J.Q.; Shaw, L.M. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013, 126, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobio. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. Amyloid-beta oligomer Aβ*56 induces specific alterations of tau phosphorylation and neuronal signaling. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Wolf, A.; Strom, A.; Wang, P.; Iaccarino, L.; Bourakova, V.; Cobigo, Y.; Heuer, H.; Spina, S.; et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med. 2020, 26, 387–397. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [Green Version]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Polymorphism in Alzheimer Abeta amyloid organization reflects conformation selection in a rugged energy landscape. Chem. Rev. 2010, 110, 4820–4838. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; & Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Wang, H.W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A.; et al. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Ladiwala, R.A.; Dordick, J.S.; Tessier, P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. J. Biol. Chem. 2011, 286, 3209–3218. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef] [Green Version]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol-Pina, R.; Vilaprinyó-Pascual, S.; Mazzucato, R.; Arcella, A.; Vilaseca, M.; Orozco, M.; Carulla, N. SDS-PAGE analysis of Aβ oligomers is disserving research into Alzheimer’s disease: Appealing for ESI-IM-MS. Sci. Rep. 2015, 5, 14809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, J.T.; Berger, E.P.; Lansbury, J.P.T. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Portelius, E.; Andreasson, U.; Ringman, J.M.; Buerger, K.; Daborg, J.; Buchhave, P.; Hansson, O.; Harmsen, A.; Gustavsson, M.K.; Hanse, E.; et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.E.; Ruotolo, B.T.; Robinson, C.V.; et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Economou, N.J.; Giammona, M.J.; Do, T.D.; Zheng, X.; Teplow, D.B.; Buratto, S.K.; Bowers, M.T. Amyloid beta-protein assembly and Alzheimer’s disease: Dodecamers of Aβ42, but not of Aβ40, seed fibril formation. J. Am. Chem. Soc. 2016, 138, 1772–1775. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers—What you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Hepler, R.W.; Grimm, K.M.; Nahas, D.D.; Breese, R.; Dodson, E.C.; Acton, P.; Keller, P.M.; Yeager, M.; Wang, H.; Shughrue, P.; et al. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry 2006, 45, 15157–15167. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Hsu, C.C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-sheet secondary structure in amyloid-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 118, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Shea, D.; Colasurdo, E.; Smith, A.; Paschall, C.; Shofer, J.B.; Jayadev, S.; Keene, C.D.; Galasko, D.; Ko, A.; Li, G.; Peskind, E.; Daggett, V. SOBA: Development and testing of a soluble oligomer binding assay for detection of amyloidogenic toxic oligomers. Proc. Natl. Acad. Sci. USA, 2022; submitted. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 5618, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, 74–78. [Google Scholar] [CrossRef]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay III, R.; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodeg. 2010, 5, 57. [Google Scholar] [CrossRef] [Green Version]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Aβ oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Emmerling, M.R.; Vigo-Pelfrey, C.; Kasunic, T.C.; Kirkpatrick, J.B.; Murdoch, G.H.; Ball, M.J.; Roher, A.E. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 1996, 271, 4077–4081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Wong, P.T.; Lee, E.L.; Gafni, A.; Steel, D.G. Determination of the oligomer size of amyloidogenic protein beta-amyloid(1-40) by single-molecule spectroscopy. Biophys. J. 2009, 97, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Kirby, N.; Mertens, H.D.T.; Roberts, B.; Barnham, K.J.; Cappai, R.; Pham, C.L.L.; Masters, C.L.; Curtain, C.C. Small angle x-ray scattering analysis of Cu2+-induced oligomers of the Alzheimer’s amyloid β peptide. Metallomics 2015, 7, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.; Ma, B.; Tsai, C.J.; Nussinov, R. Hollow core of Alzheimer’s Aβ42 amyloid observed by cryoEM is relevant at physiological pH. Proc. Natl. Acad. Sci. USA 2010, 107, 14128–14133. [Google Scholar] [CrossRef] [Green Version]
- Hyung, S.J.; DeToma, A.S.; Brender, J.R.; Lee, S.; Vivekanandan, S.; Kochi, A.; Choi, J.S.; Ramamoorthy, A.; Ruotolo, B.T.; Lim, M.H. Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-β species. Proc. Natl. Acad. Sci. USA 2013, 110, 3743–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrick, J.S.; Kerr, R.A.; Nam, Y.; Oh, S.B.; Lee, H.J.; Earnest, K.G.; Suh, N.; Peck, K.L.; Ozbil, M.; Korshavn, K.J.; et al. A redox-active, compact molecule for cross-linking amyloidogenic peptides into nontoxic, off-pathway aggregates: In vitro and in vivo efficacy and molecular mechanisms. Am. Chem. Soc. 2015, 137, 14785–14797. [Google Scholar] [CrossRef] [Green Version]
- Armen, R.; Alonso, D.; Daggett, V. Anatomy of an amyloidgenic intermediate: Conversion of β-sheet to α-pleated sheet structure in transthyretin at acidic pH. Structure 2004, 12, 1847–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armen, R.; DeMarco, M.; Alonso, D.; Daggett, V. Pauling and Corey’s α-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA 2004, 101, 11622–11627. [Google Scholar] [CrossRef] [Green Version]
- Daggett, V. α-sheet: The toxic conformer in amyloid diseases? Acc. Chem. Res. 2006, 39, 594–602. [Google Scholar] [CrossRef]
- Hopping, G.; Kellock, J.; Barnwal, R.P.; Law, P.; Bryers, J.; Varani, G.; Caughey, B.; Daggett, V. Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. eLife 2014, 3, e01681. [Google Scholar] [CrossRef]
- Kellock, J.; Hopping, G.; Caughey, B.; Daggett, V. Peptides composed of alternating L- and D-amino acids inhibit amyloidogenesis in three distinct amyloid systems independent of sequence. J. Mol. Biol. 2016, 428, 2317–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, N.; Shea, D.; Bleem, A.; Bryers, J.D.; Daggett, V. Chemical and physical variability in structural isomers of an L/D α-sheet peptide designed to inhibit amyloidogenesis. Biochemistry 2018, 57, 507–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleem, A.; Francisco, R.; Bryers, J.D.; Daggett, V. Designed alpha-sheet peptides suppress amyloid formation in Staphylococcus aureus biofilms. Nat. Biofilms Microbiomes 2017, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, T.M.; Daggett, V. The role of alpha-sheet in amyloid oligomer aggregation and toxicity. Yale J. Biol. Med. 2018, 91, 247–255. [Google Scholar]
- Daggett, V.; Fersht, A. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 2003, 4, 497–502. [Google Scholar] [CrossRef]
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.K.; Ramamoorthy, A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Chimon, S.; Shaibat, M.A.; Jones, C.R.; Calero, D.C.; Aizezi, B.; Ishii, Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007, 14, 1157–1164. [Google Scholar] [CrossRef]
- Pham, J.D.; Chim, N.; Goulding, C.W.; Nowick, J.S. Structures of oligomers of a peptide from β-amyloid. J. Am. Chem. Soc. 2013, 135, 12460–12467. [Google Scholar] [CrossRef] [Green Version]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure Alzheimer’s beta-amyloid fibrils. Biochem. 2006, 45, 498–512. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Ideno, S.; et al. Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology and toxicity analyses. J. Biol. Chem. 2011, 286, 11555–11562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanpaa, K.; Isaacs, K.R.; Shirao, T.; Brady, D.R.; Rapoport, S.I. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropath. Exp. Neur. 1999, 58, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [Green Version]
- Cerf, E.; Sarroukh, R.; Tamamizu-Kato, S.; Breydo, L.; Derclaye, S.; Dufrene, Y.F.; Narayanaswami, V.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. Antiparallel beta-sheet: A signature structure of the oligomeric amyloid beta-peptide. Biochem. J. 2009, 421, 415–423. [Google Scholar] [CrossRef]
- Fonte, V.; Dostal, V.; Roberts, C.M.; Gonzales, P.; Lacor, P.; Magrane, J.; Dingwell, N.; Fan, E.Y.; Silverman, M.A.; Stein, G.H.; et al. A glycine zipper motif mediates the formation of toxic β-amyloid oligomers in vitro and in vivo. Mol. Neurodeg. 2011, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.W.; Ciccotosto, G.D.; Giannakis, E.; Tew, D.J.; Perez, K.; Masters, C.L.; Cappai, R.; Wade, J.D.; Barnham, K.J. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neuro. 2008, 28, 11950–11958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, 2518–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; McLaurin, J. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alz. Dis. 2010, 2011, 548380. [Google Scholar] [CrossRef] [Green Version]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; de Oliveira, C.R. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1–42: Involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 2008, 155, 725–737. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Wogulis, M.; Wright, S.; Cunningham, D.; Chilcote, T.; Powell, K.; Rydel, R.E. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J. Neurosci. 2005, 25, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; Garcia-Vinuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-chaperone hypothesis: A common molecular mechanism of membrane disruption by intrinsically disordered proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alz. Dem. Trans. Res. Clin. Int. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Haeberlein, S.H.; O’Gorman, J.; Chiao, P.; Bussiere, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical development of Aducanumab, an anti-Aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J. Prev. Alz. Dis. 2017, 4, 255–263. [Google Scholar]
- Imbimbo, B.P.; Ottonello, S.; Frisardi, V.; Solfrizzi, V.; Greco, A.; Seripa, D.; Pilotto, A.; Panza, F. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev. Clin. Immunol. 2012, 8, 135–149. [Google Scholar] [CrossRef]
- Samadi, H.; Sultzer, D. Solanezumab for Alzheimer’s disease. Drug Eval. 2011, 11, 787–798. [Google Scholar] [CrossRef]
- Meilandt, W.; Maloney, J.; Imperio, J.; Bainbridge, T.; Reichelt, M.; Mandikian, D.; Lu, Y.; Ernst, J.; Fuji, R.; Atwal, J. Characterization of the selective in vivo and in vitro binding properties of Crenezumab: Insights into Crenezumab’s unique mechanism of action. Neurology 2018, 90, 174. [Google Scholar]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018, 90, 1889–1897. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neuro. 2012, 32, 9677–9689. [Google Scholar] [CrossRef]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C.; et al. BACE1 inhibitor Lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. J. Clin. Pharmacol. 2017, 57, 1460–1471. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pathology of disease progression and characterization techniques. Low molecular weight aggregates are soluble and contain primarily α-sheet structure that have been characterized by size and structure by the associated techniques. High molecular weight aggregates are soluble but contain primarily β-sheet structure that can be characterized by the associated techniques.
Figure 1.
Pathology of disease progression and characterization techniques. Low molecular weight aggregates are soluble and contain primarily α-sheet structure that have been characterized by size and structure by the associated techniques. High molecular weight aggregates are soluble but contain primarily β-sheet structure that can be characterized by the associated techniques.

Figure 2.
Isolation techniques and aggregate characteristics of toxicity and structure. Arrows span the molecular weight range that each technique can be used to characterize. Dark blue indicates largely unstructured, random coil species, red indicates assemblies that have been determined as the α-sheet-containing primary toxic aggregates, and gray indicates the HMW β-sheet aggregates and fibrils. The very LMW aggregates are soluble, unstructured, and largely nontoxic, while the intermediate 6–12 mers contain α-sheet and are the most toxic. HMW oligomers and fibrils contain β-sheet and are nontoxic.
Figure 2.
Isolation techniques and aggregate characteristics of toxicity and structure. Arrows span the molecular weight range that each technique can be used to characterize. Dark blue indicates largely unstructured, random coil species, red indicates assemblies that have been determined as the α-sheet-containing primary toxic aggregates, and gray indicates the HMW β-sheet aggregates and fibrils. The very LMW aggregates are soluble, unstructured, and largely nontoxic, while the intermediate 6–12 mers contain α-sheet and are the most toxic. HMW oligomers and fibrils contain β-sheet and are nontoxic.

Figure 3.
Drug candidates and their associated targets. Processing of APP by β-secretase produces monomeric, native Aβ. Aβ undergoes a misfolding event that eventually produces LMW oligomers that are neurotoxic and initiate the oligomer toxicity cascade. These eventually progress to HMW protofibrils that finally deposit as fibril plaques at the neuronal membrane. Lanabecestat™ is a BACE inhibitor that reduces processing of APP to produce Aβ monomer, while Solanezumab™ is a monomer (sequence) specific drug. Crenezumab™ specifically targets the β-sheet rich HMW protofibrils and fibrillar plaque deposits, while Aducanumab™ specifically targets the β-sheet fibril plaques.
Figure 3.
Drug candidates and their associated targets. Processing of APP by β-secretase produces monomeric, native Aβ. Aβ undergoes a misfolding event that eventually produces LMW oligomers that are neurotoxic and initiate the oligomer toxicity cascade. These eventually progress to HMW protofibrils that finally deposit as fibril plaques at the neuronal membrane. Lanabecestat™ is a BACE inhibitor that reduces processing of APP to produce Aβ monomer, while Solanezumab™ is a monomer (sequence) specific drug. Crenezumab™ specifically targets the β-sheet rich HMW protofibrils and fibrillar plaque deposits, while Aducanumab™ specifically targets the β-sheet fibril plaques.


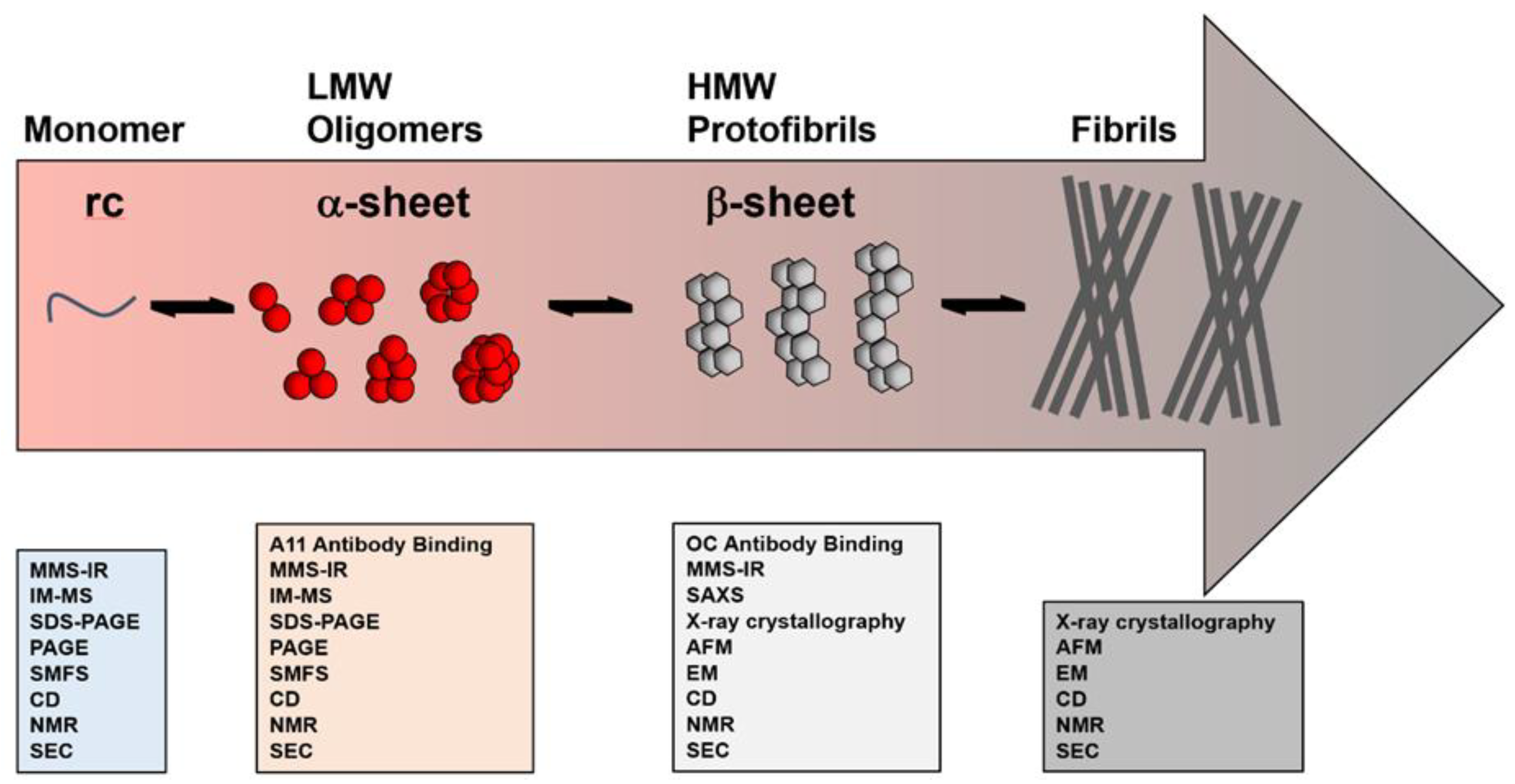{kind=link}
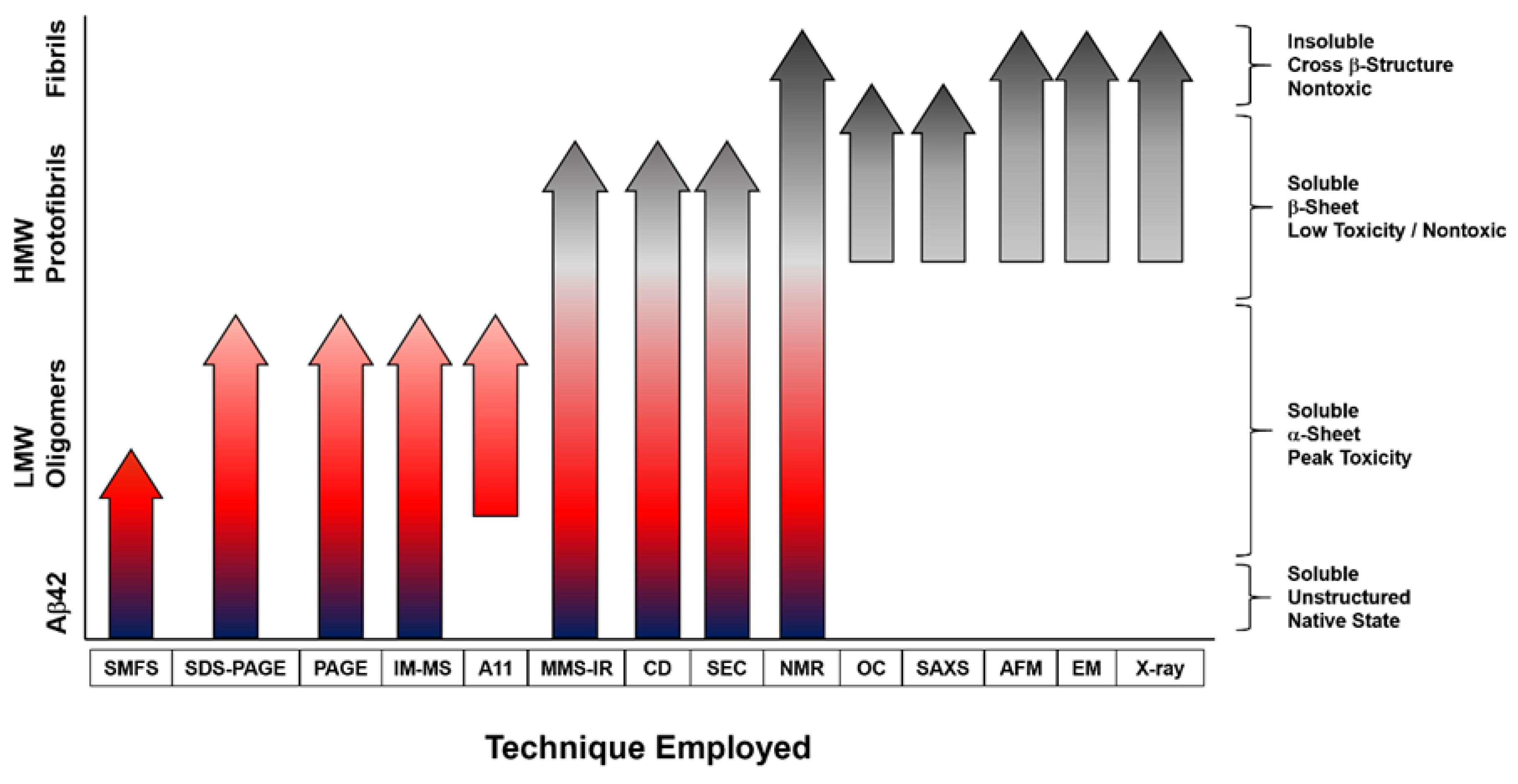{kind=link}
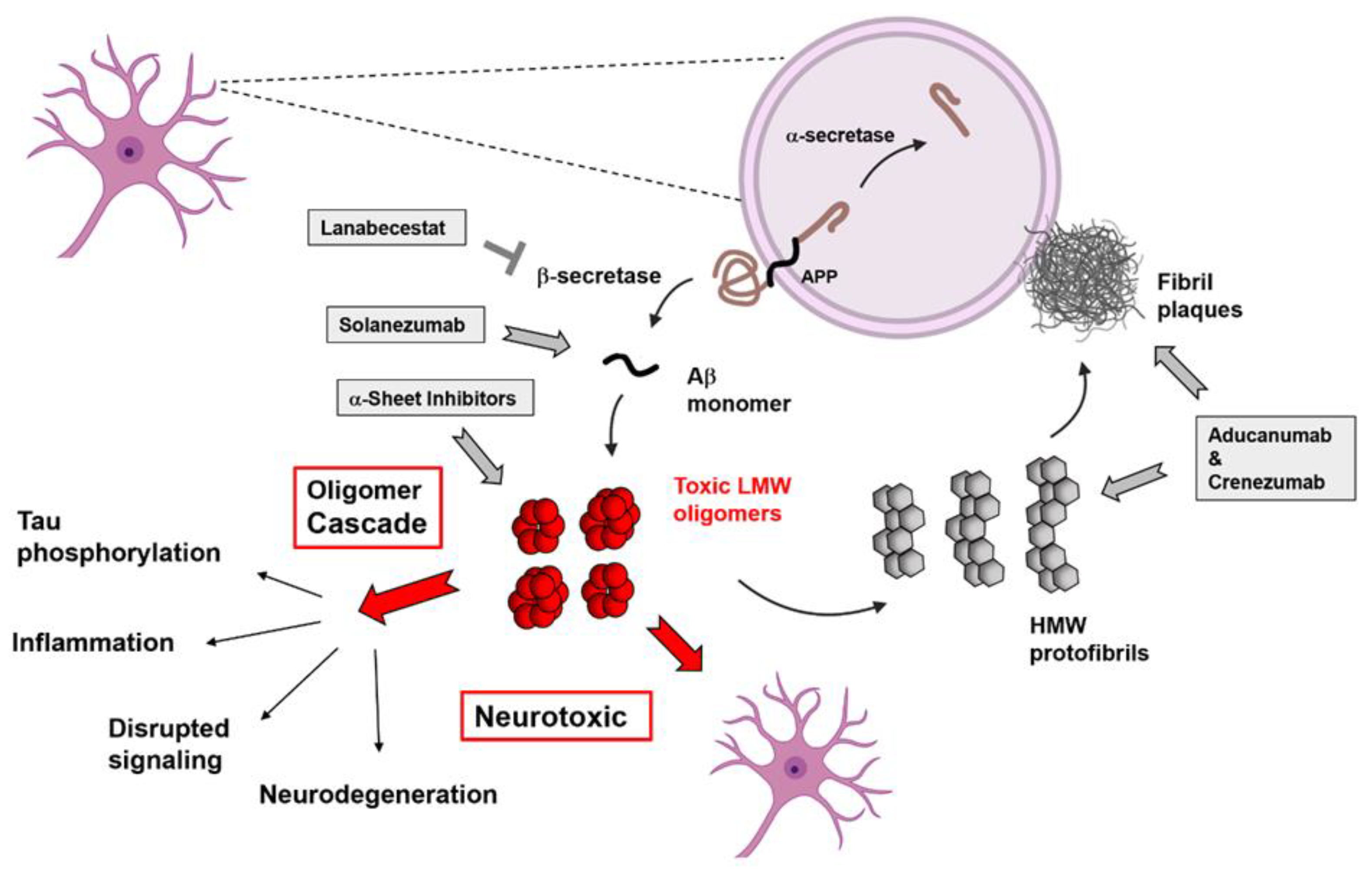{kind=link}
Table 1.
Aβ oligomers observed in different studies and by different techniques.
| Species | Size & Characterizations | Isolation Technique (Source, Ie In Vitro) |
|---|---|---|
| SDS-stable dimers and synthetic dimers [65] | Dimers, 8–12 kDa, 3–4 nm height, no detected secondary structure | SDS-PAGE (brain derived or synthetic in vitro Aβ40Ser26Cys mutant) |
| SDS-stable dimers/trimers [67] | Dimer/trimer, 6–12 kDa, Aβ40/Aβ42 with Arg5 N-term truncated species | SDS-PAGE (transfected CHO cell culture medium) |
| Trimers from mutant human APPV717F [64] | Trimers, 12 kDa, unstructured | SEC and PAGE (7PA2 cells) |
| Tetramers (Aβ42 and Aβ40) [80] | Tetramer, 18 kDa, ring-shaped (Aβ40) or bent (Aβ42), non-aggregation prone (Aβ40) or aggregation prone (Aβ42) | IM-MS (synthetic in vitro) |
| Pentamer [22] | Pentamer, compact pentagonal shape with C-termini buried | ssNMR (synthetic in vitro kinetically trapped) |
| Hexamer/dodecamer [85,86] | Hexamer-dodecamer, 27–56 kDa, α-sheet secondary structure, A11 positive | SEC (synthetic in vitro, human CSF) |
| Aβ*56 [63] | Dodecamer, ~56 kDa, globular, A11 positive | SEC and PAGE (Human brain isolates) |
| Aβo [87,88,89] | 15–20 mer, spherical vesicles 2–5 nm diameter, A11 positive | SEC (synthetic in vitro) |
| ADDLs [21] | Trimer-24 mer, 17 kDa tetramer major, globular, 2–5 nm height, A11-positive | Nondenaturing electrophoresis (synthetic in vitro) |
| ASPD (amylospheroids) [90] | 32–150 mers, spheroids, 10–15 nm diameter assemblies, A11 negative | SDS-PAGE (brain derived and synthetic in vitro) |
| Aβ42 Ellipsoids [66] | High molecular weight, ellipsoidal and annular | SAXS (Cu(II)-guided Aβ42 oligomerization in vitro) |
| Aβ40 Protofibrils [61] | High molecular weight, protofibrillar | SAXS (Cu(II)-guided Aβ40 oligomerization in vitro) |
| HMW soluble oligomers [66] | Large, circular, 8–12 nm, form membrane-permeable pore at lipid bilayers | AFM (synthetic in vitro) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shea, D.; Daggett, V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica 2022, 2, 91-110. https://doi.org/10.3390/biophysica2020010
AMA Style
Shea D, Daggett V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica. 2022; 2(2):91-110. https://doi.org/10.3390/biophysica2020010
Chicago/Turabian StyleShea, Dylan, and Valerie Daggett. 2022. "Amyloid-β Oligomers: Multiple Moving Targets" Biophysica 2, no. 2: 91-110. https://doi.org/10.3390/biophysica2020010