Molecular Forces Governing the Biological Function of Per-Arnt-Sim-B (PAS-B) Domains: A Comparative Computational Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Homology Modelling
2.2. Molecular Dynamics Simulations
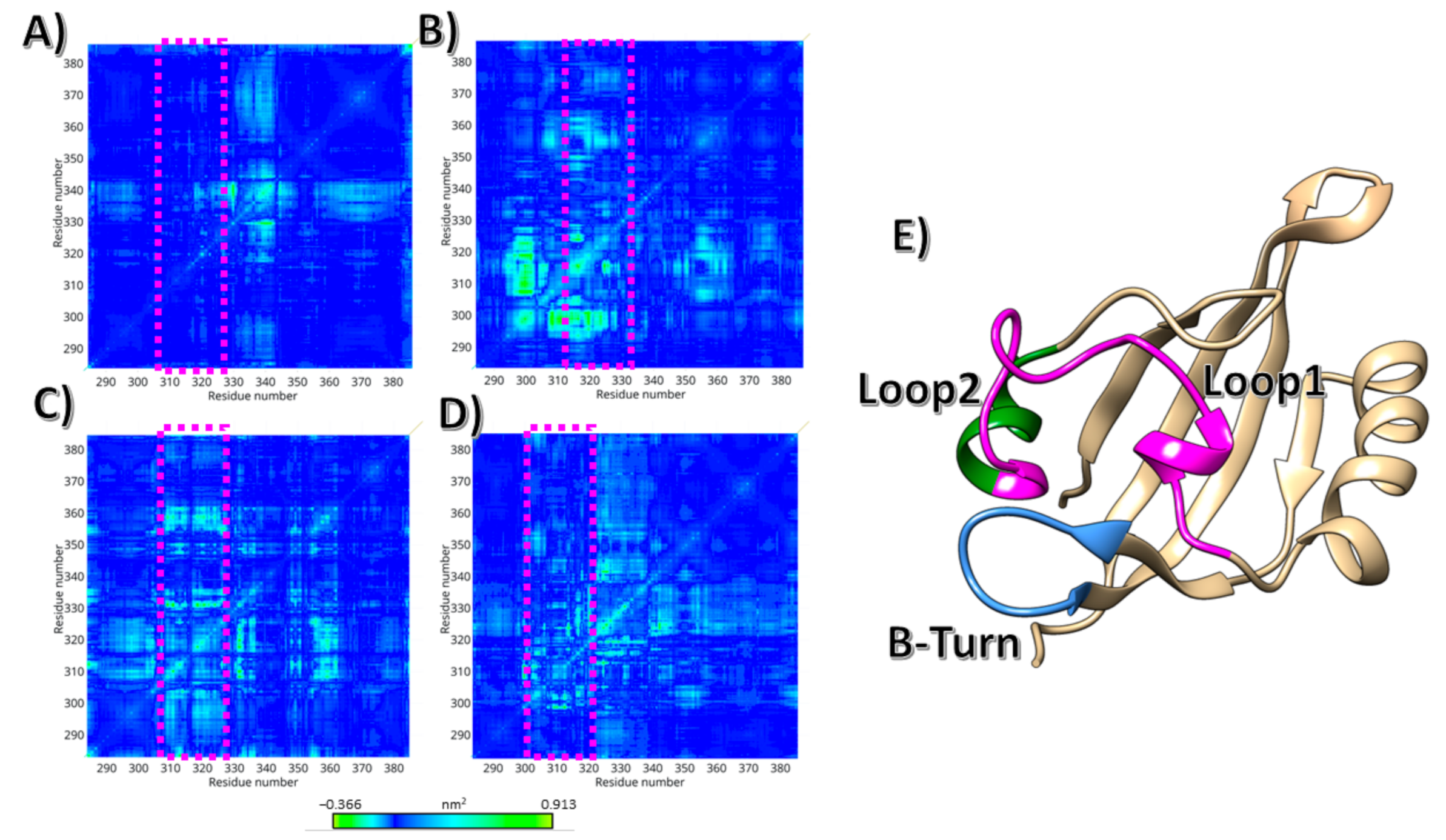
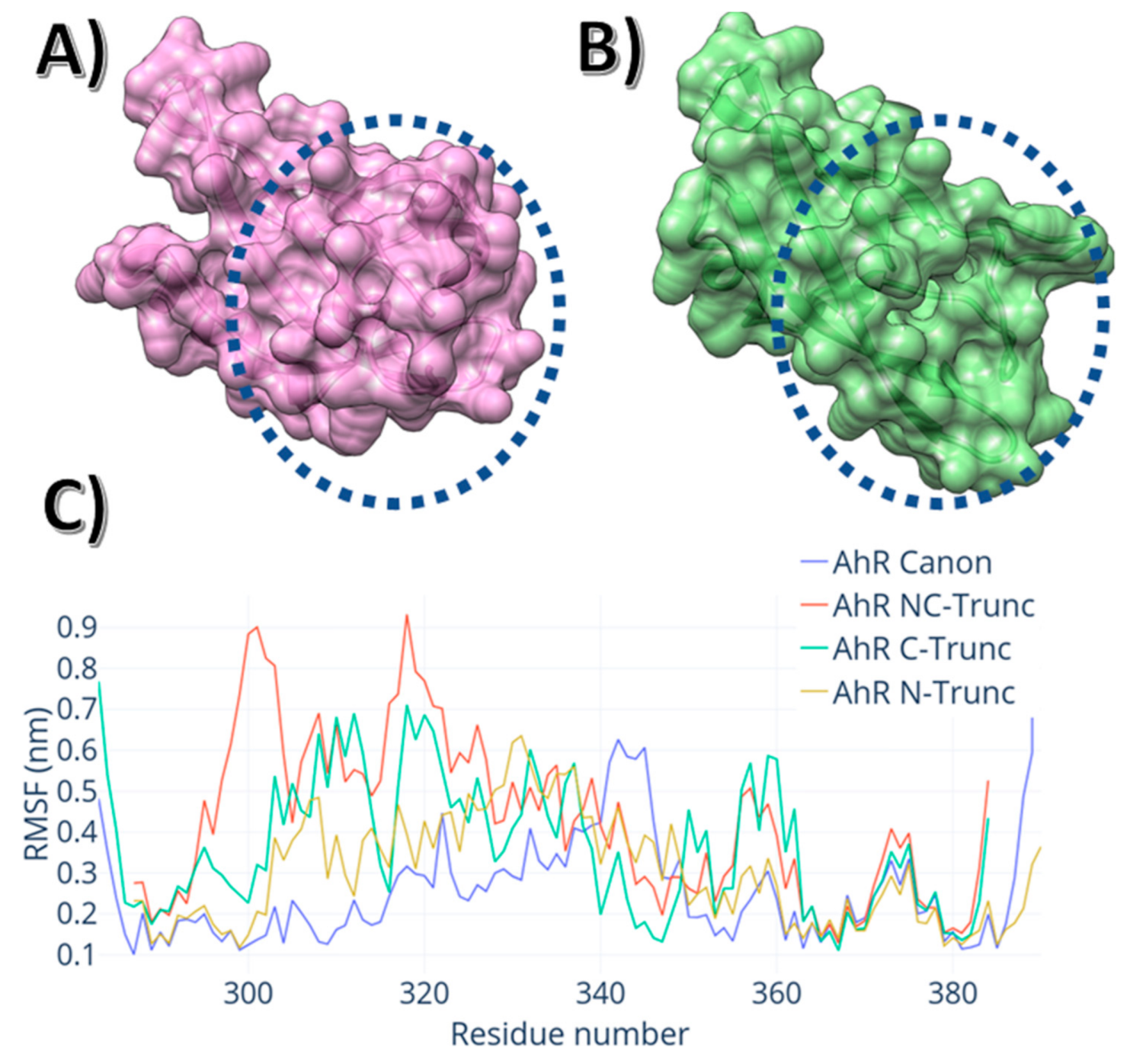
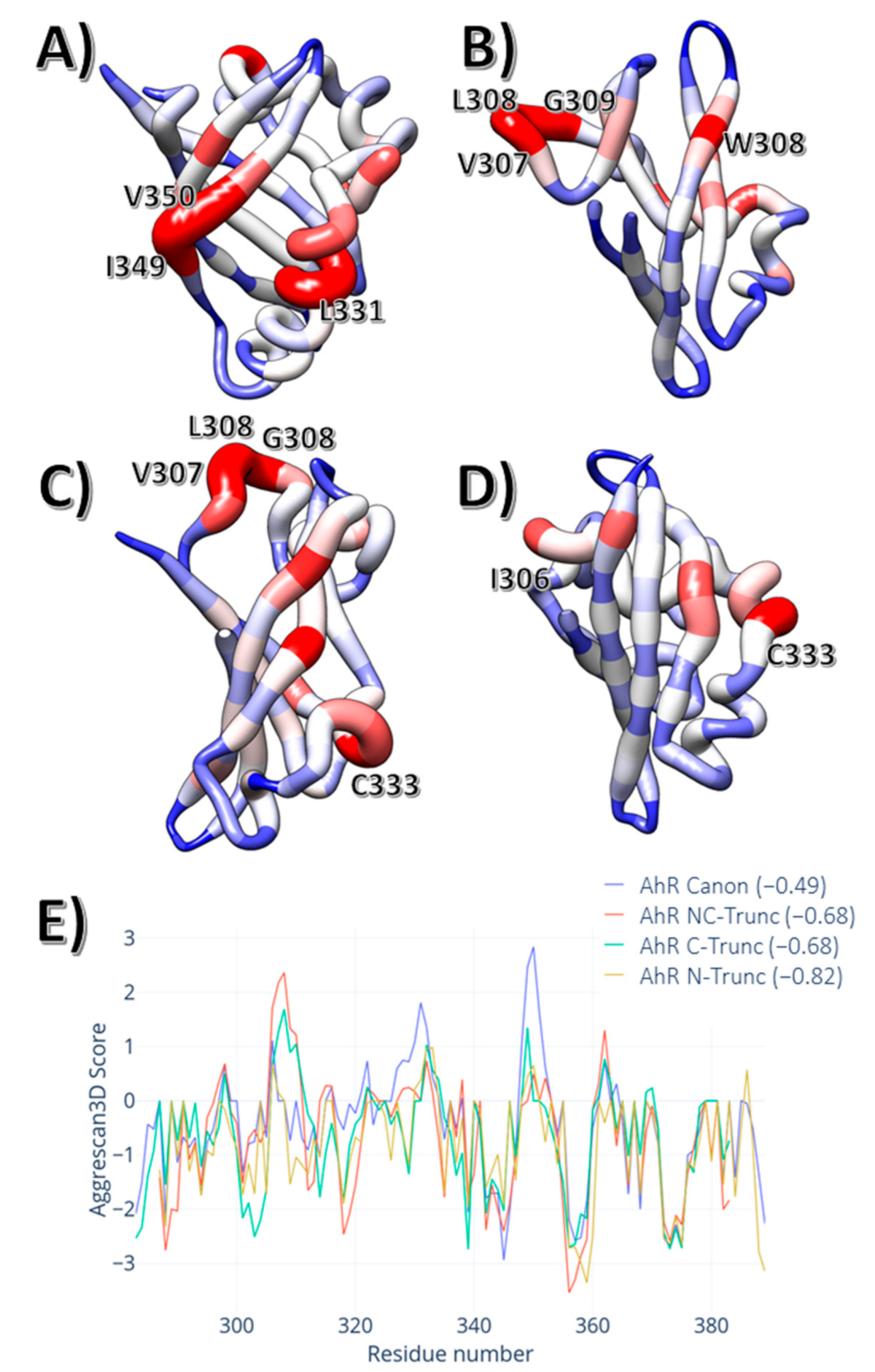
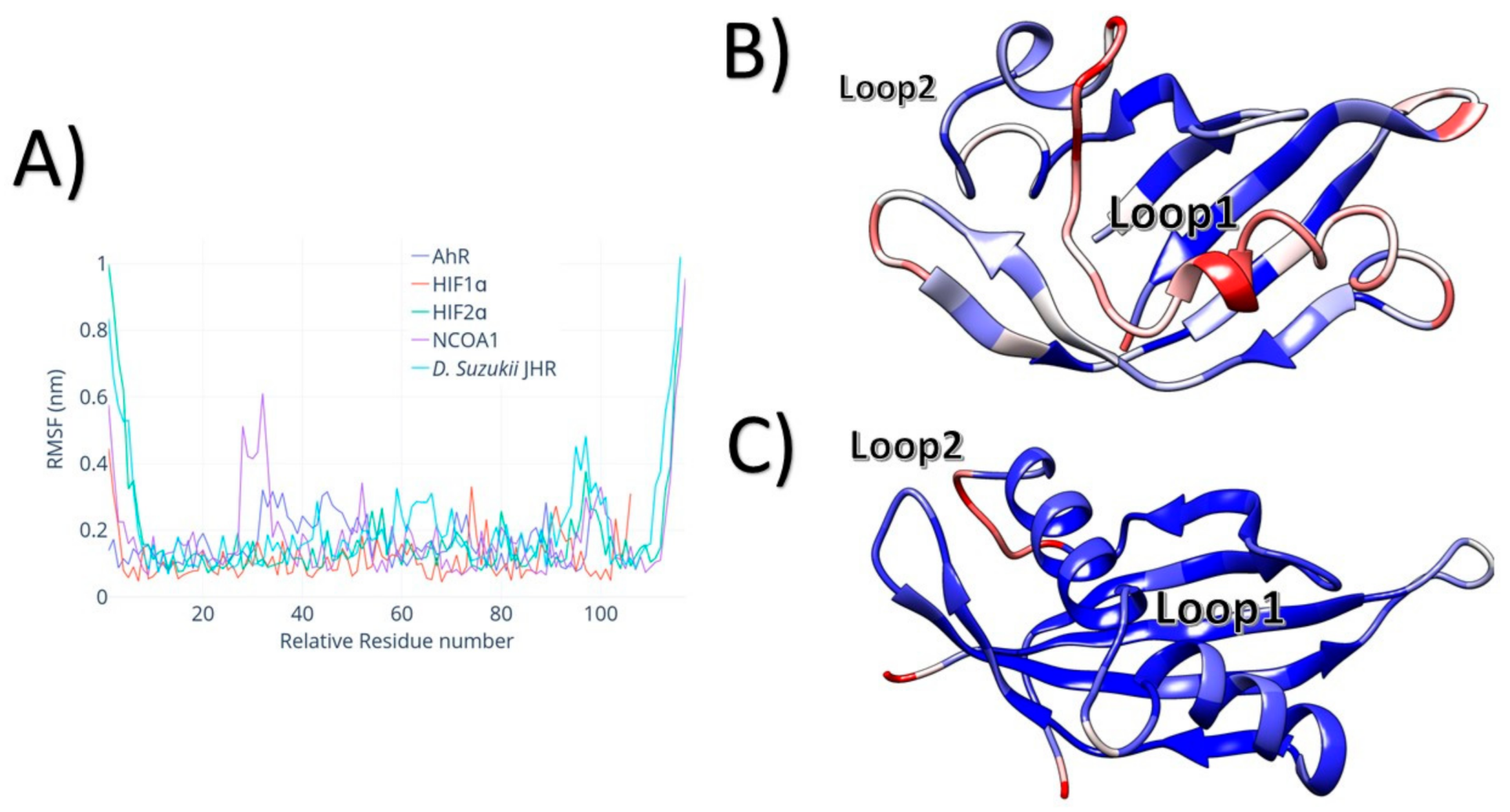
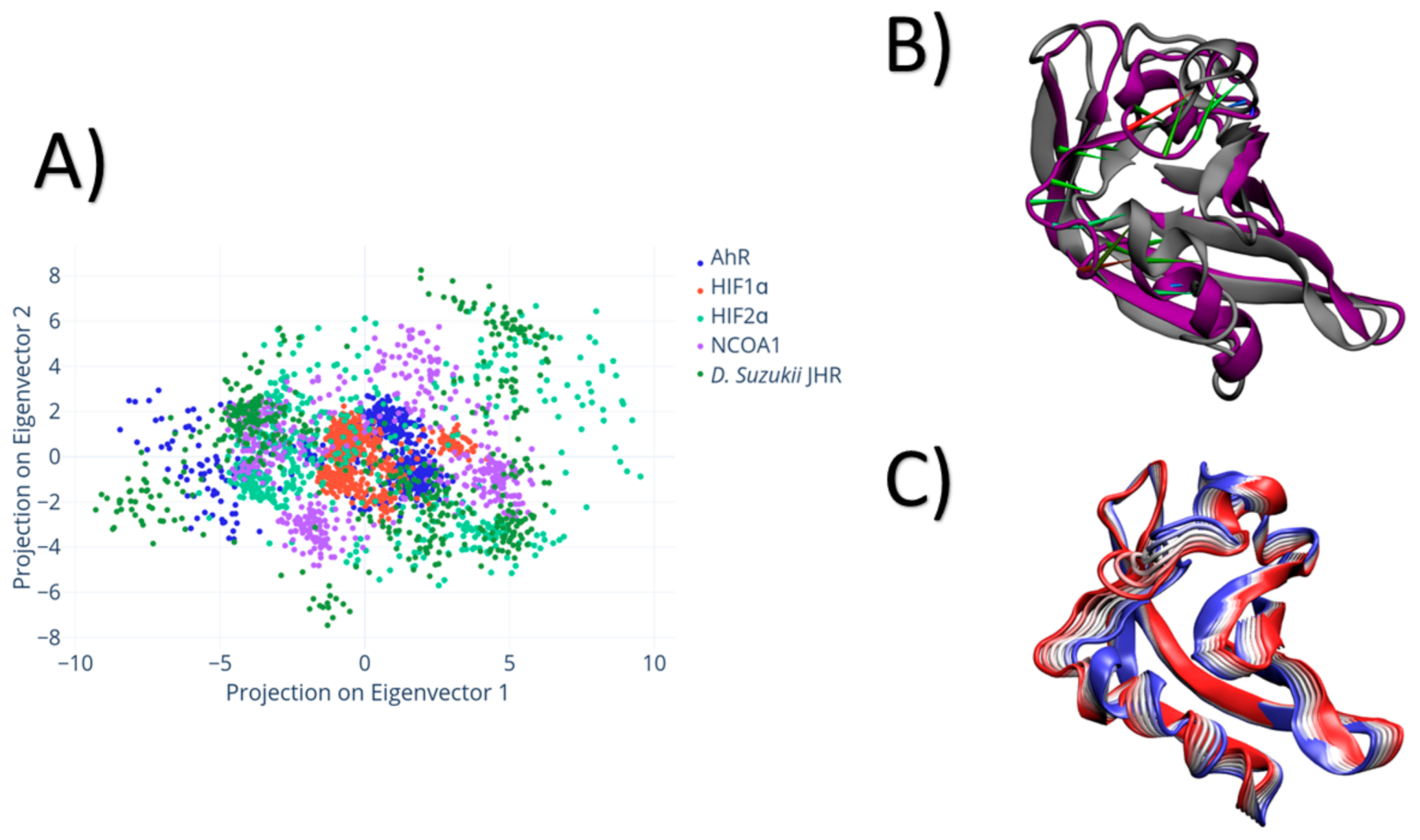
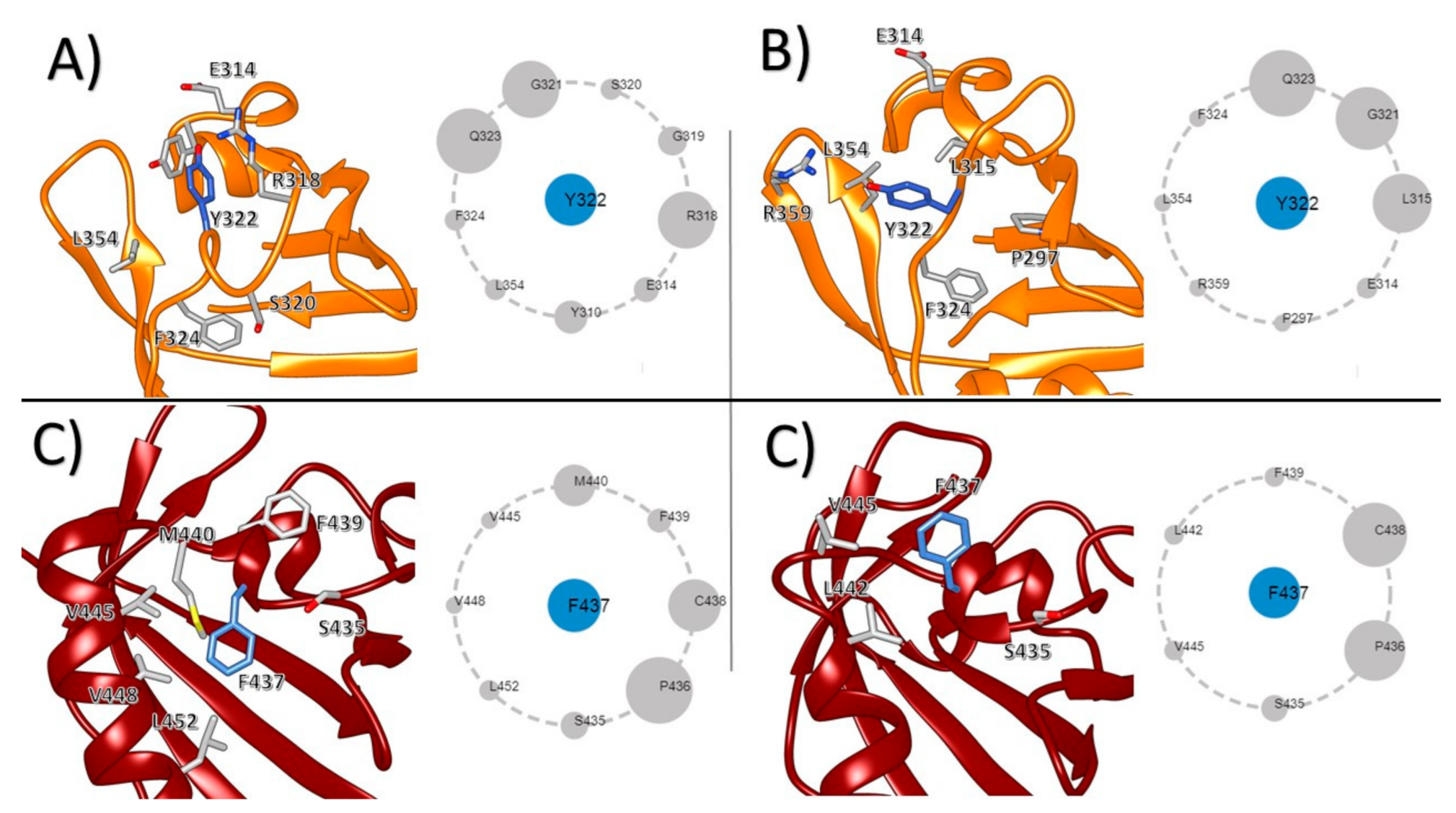
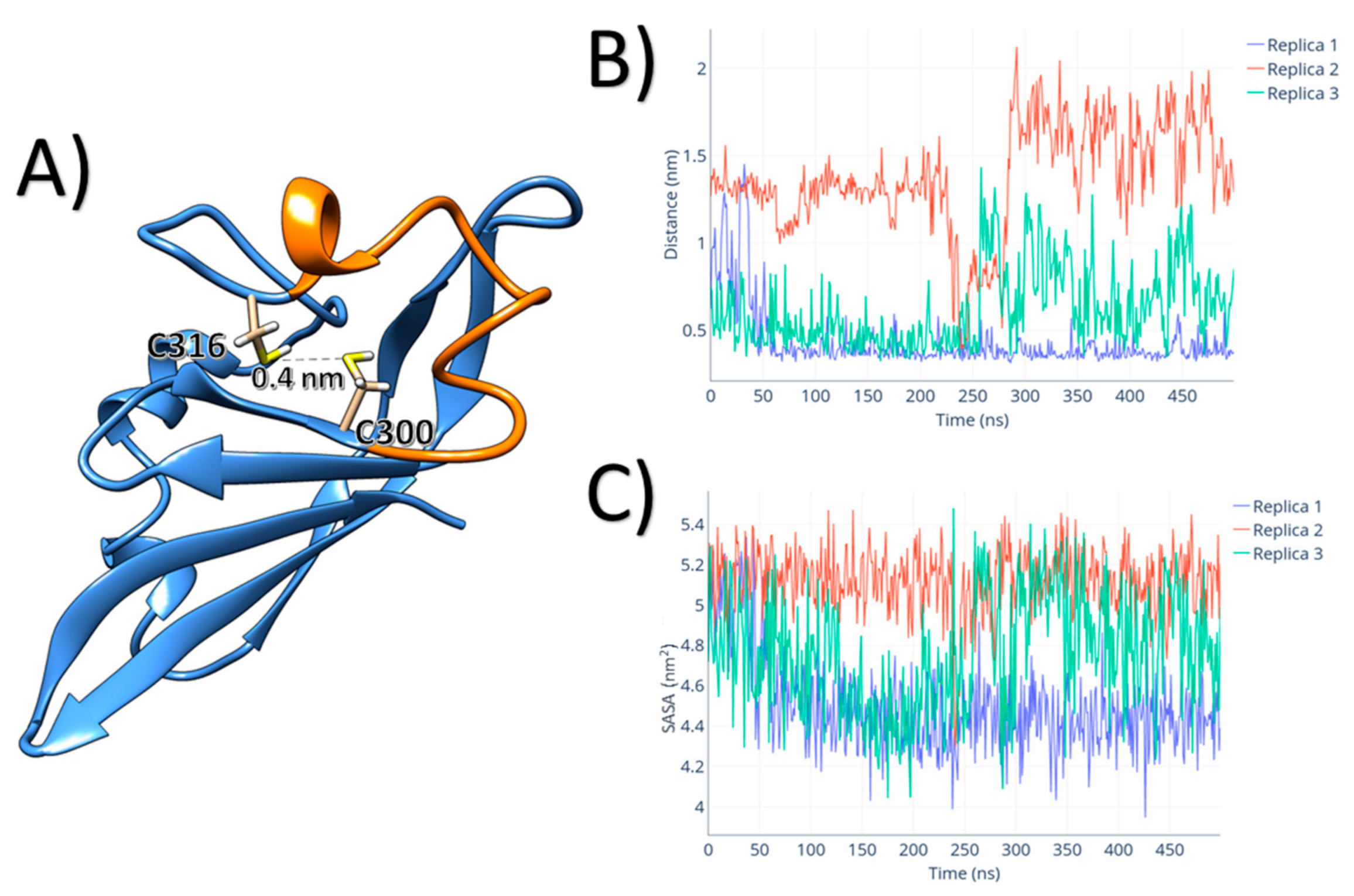
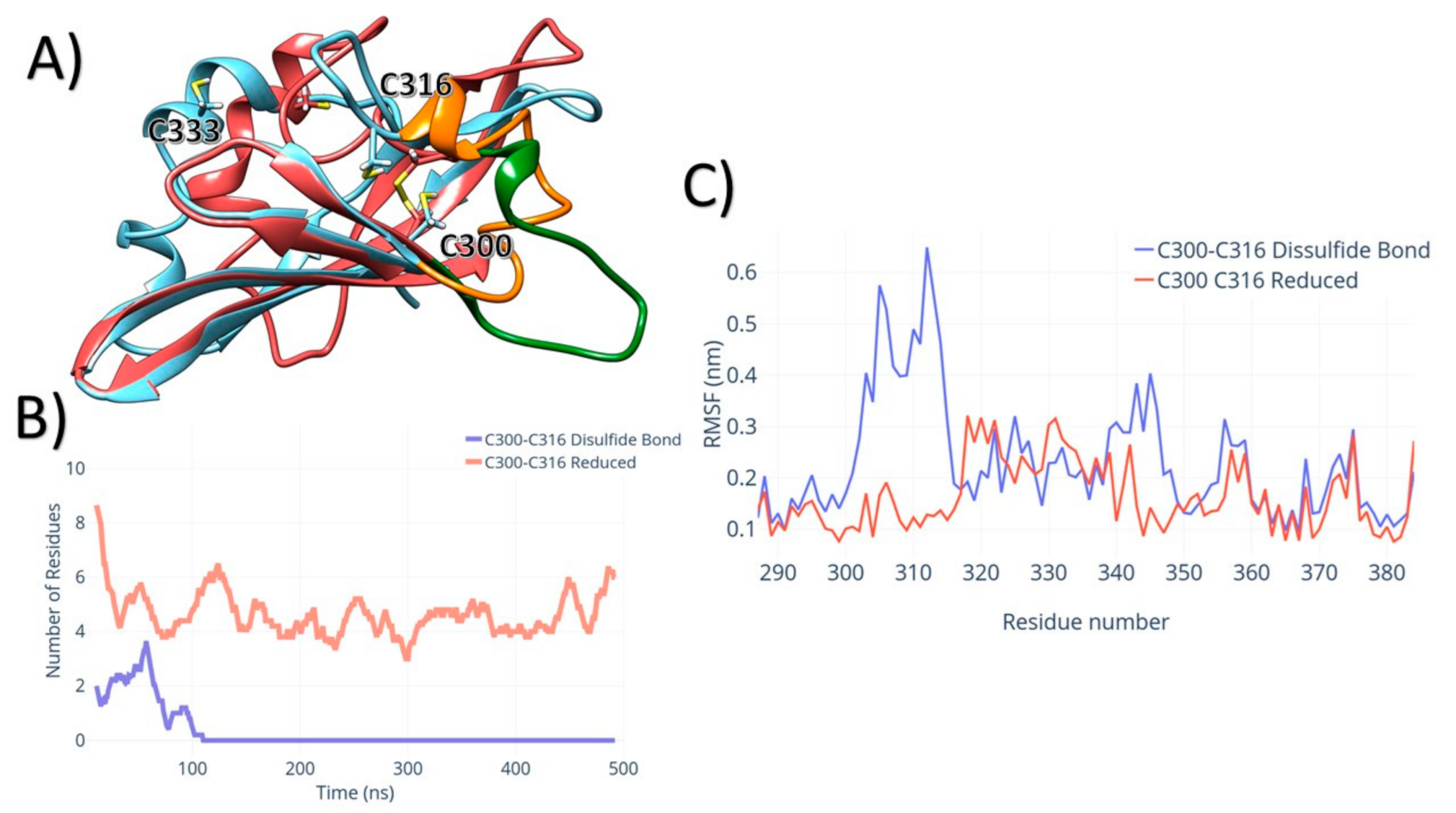
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, Z.; Zou, X.; Chen, Y.; Wang, H.; Duan, Y.; Bruick, R.K. Modulation of HIF-2α PAS-B domain contributes to physiological responses. Proc. Natl. Acad. Sci. USA 2018, 115, 13240–13245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depping, R.; Oster, H. Interplay between environmentally modulated feedback loops—hypoxia and circadian rhythms—two sides of the same coin? FEBS J. 2017, 284, 3801–3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z.; Santer, F.R. Studies on Steroid Receptor Coactivators in Prostate Cancer. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1786, pp. 259–262. [Google Scholar]
- Zhang, Y.; Markert, M.J.; Groves, S.C.; Hardin, P.E.; Merlin, C. Vertebrate-like CRYPTOCHROME 2 from monarch regulates circadian transcription via independent repression of CLOCK and BMAL1 activity. Proc. Natl. Acad. Sci. USA 2017, 114, E7516–E7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Pirela, M.; Rigden, D.J.; Michels, P.A.; Cáceres, A.J.; Concepción, J.L.; Quiñones, W. Structure and function of Per-ARNT-Sim domains and their possible role in the life-cycle biology of Trypanosoma cruzi. Mol. Biochem. Parasitol. 2018, 219, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Hartzell, A.L.; Martyniuk, K.M.; Brigidi, G.S.; Heinz, D.A.; Djaja, N.A.; Payne, A.; Bloodgood, B.L. NPAS4 recruits CCK basket cell synapses and enhances cannabinoid-sensitive inhibition in the mouse hippocampus. Elife 2018, 7. [Google Scholar] [CrossRef]
- Harmon, A.C.; Hebert, V.Y.; Cormier, S.A.; Subramanian, B.; Reed, J.R.; Backes, W.L.; Dugas, T.R. Particulate matter containing environmentally persistent free radicals induces AhRdependent cytokine and reactive oxygen species production in human bronchial epithelial cells. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Su, X.; Potluri, N.; Kim, Y.; Rastinejad, F. NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH-PAS family as multi-ligand binding transcription factors. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- De Souza, J.V.; Reznikov, S.; Zhu, R.; Bronowska, A.K. Druggability assessment of mammalian Per-Arnt-Sim [PAS] domains using computational approaches. Medchemcomm 2019, 10, 1126–1137. [Google Scholar] [CrossRef]
- Weng, F.J.; Garcia, R.I.; Lutzu, S.; Alviña, K.; Zhang, Y.; Dushko, M.; Ku, T.; Zemoura, K.; Rich, D.; Garcia-Dominguez, D.; et al. Npas4 Is a Critical Regulator of Learning-Induced Plasticity at Mossy Fiber-CA3 Synapses during Contextual Memory Formation. Neuron 2018, 97, 1137–1152.e5. [Google Scholar] [CrossRef] [Green Version]
- Shinde, R.; McGaha, T.L. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends Immunol. 2018, 39, 1005–1020. [Google Scholar] [CrossRef]
- Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. J. Clin. Med. 2018, 7, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl sulfate affects glial function increasing oxidative stress and neuroinflammation in chronic kidney disease: Interaction between astrocytes and microglia. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.A.; Denison, M.S. Ligand Promiscuity of Aryl Hydrocarbon Receptor Agonists and Antagonists Revealed by Site-Directed Mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolonko, M.; Greb-Markiewicz, B. BHLH–PAS proteins: Their structure and intrinsic disorder. Int. J. Mol. Sci. 2019, 20, 3653. [Google Scholar] [CrossRef] [Green Version]
- Partch, C.L.; Gardner, K.H. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc. Natl. Acad. Sci. USA 2011, 108, 7739–7744. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Toyota, K.; Ishibashi, H.; Tominaga, N.; Sato, T.; Tatarazako, N.; Iguchi, T. Molecular Insights into Structural and Ligand Binding Features of Methoprene-Tolerant in Daphnids. Chem. Res. Toxicol. 2020. [Google Scholar] [CrossRef]
- Charles, J.P.; Iwema, T.; Epa, V.C.; Takaki, K.; Rynes, J.; Jindra, M. Ligand-binding properties of a juvenile hormone receptor, Methoprene-tolerant. Proc. Natl. Acad. Sci. USA 2011, 108, 21128–21133. [Google Scholar] [CrossRef] [Green Version]
- Dahm, K.H.; Bhaskaran, G.; Peter, M.G.; Shirk, P.D.; Seshan, K.R.; Röller, H. On the Identity of the Juvenile Hormone in Insects. In The Juvenile Hormones; Springer: New York, NY, USA, 1976; pp. 19–47. [Google Scholar]
- Jindra, M.; Uhlirova, M.; Charles, J.-P.; Smykal, V.; Hill, R.J. Genetic Evidence for Function of the bHLH-PAS Protein Gce/Met As a Juvenile Hormone Receptor. PLoS Genet. 2015, 11, e1005394. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Russo, L.; Giller, K.; Pfitzner, E.; Griesinger, C.; Becker, S. Insight into the molecular recognition mechanism of the coactivator NCoA1 by STAT6. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, D.R.; Poland, A. Binding of aryl hydrocarbon receptor (AhR) to AhR-interacting protein: The role of hsp90. J. Biol. Chem. 2000, 275, 36407–36414. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, N.; Fukuda, K.; Nagata, Y.; Okada, H.; Haga, A.; Hatakeyama, S.; Yoshida, S.; Okamoto, T.; Hosaka, M.; Sekine, K.; et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 2014, 4, 796–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, E.C.; Gasiewicz, T.A. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. J. 1993, 294, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, B.N.; Probst, M.R.; Reisz-Porszasz, S.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 1995, 270, 29270–29278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keul, N.D.; Oruganty, K.; Bergman, E.T.S.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The entropic force generated by intrinsically disordered segments tunes protein function. Nature 2018, 563, 584–588. [Google Scholar] [CrossRef]
- Jin, H.; Ji, C.; Ren, F.; Aniagu, S.; Tong, J.; Jiang, Y.; Chen, T. AHR-mediated oxidative stress contributes to the cardiac developmental toxicity of trichloroethylene in zebrafish embryos. J. Hazard. Mater. 2020, 385. [Google Scholar] [CrossRef] [PubMed]
- Omidi, M.; Ghafarian-Bahraman, A.; Mohammadi-Bardbori, A. GSH/GSSG redox couple plays central role in aryl hydrocarbon receptor-dependent modulation of cytochrome P450 1A1. J. Biochem. Mol. Toxicol. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Kuriata, A.; Iglesias, V.; Pujols, J.; Kurcinski, M.; Kmiecik, S.; Ventura, S. Aggrescan3D (A3D) 2.0: Prediction and engineering of protein solubility. Nucleic Acids Res. 2019, 47, W300–W307. [Google Scholar] [CrossRef] [Green Version]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef]
- Soylu, İ.; Marino, S.M. Cy-preds: An algorithm and a web service for the analysis and prediction of cysteine reactivity. Proteins Struct. Funct. Bioinform. 2016, 84, 278–291. [Google Scholar] [CrossRef]
- Soshilov, A.; Denison, M.S. Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J. Biol. Chem. 2008, 283, 32995–33005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrada, D.; Denison, M.S.; Bonati, L. Structural modeling of the AhR:ARNT complex in the bHLH-PASA-PASB region elucidates the key determinants of dimerization. Mol. Biosyst. 2017, 13, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, J.V.; Zaborniak, P.; Reznikov, S.; Kondal, M.; Zhu, R.; Bronowska, A.K. Molecular Forces Governing the Biological Function of Per-Arnt-Sim-B (PAS-B) Domains: A Comparative Computational Study. Biophysica 2021, 1, 1-14. https://doi.org/10.3390/biophysica1010001
de Souza JV, Zaborniak P, Reznikov S, Kondal M, Zhu R, Bronowska AK. Molecular Forces Governing the Biological Function of Per-Arnt-Sim-B (PAS-B) Domains: A Comparative Computational Study. Biophysica. 2021; 1(1):1-14. https://doi.org/10.3390/biophysica1010001
Chicago/Turabian Stylede Souza, João Victor, Piotr Zaborniak, Sylvia Reznikov, Matthew Kondal, Ruidi Zhu, and Agnieszka K. Bronowska. 2021. "Molecular Forces Governing the Biological Function of Per-Arnt-Sim-B (PAS-B) Domains: A Comparative Computational Study" Biophysica 1, no. 1: 1-14. https://doi.org/10.3390/biophysica1010001