
On the Genesis of a Catalyst: A Brief Review with an Experimental Case Study

Abstract
:1. Introduction
2. Genesis of a Catalyst
2.1. Selection of the Support
- (a)
- It should withstand high temperature and pressure with minimal sintering;
- (b)
- It should have an adequate texture; that is, sufficient surface area to lodge the necessary oxide particles that will play the role of the active species. It should also have adequately sized pores to facilitate diffusion of the reactants through the pores and the ability for the product to diffuse out of the pore [34,35];
- (c)
- It must have a certain surface acidity/basicity when required by the reaction;
- (d)
- Other interesting characteristics must be sometimes required such as morphology or any structure arrangement that yields to stabilize the active species.
2.2. Selection of the Metal Active Sites
2.3. Preparation of the Catalyst
- (a)
- Precipitation method: This method is sometimes known as co-precipitation and is one of the most widely used catalyst preparation methods. This method can be used to prepare a single component catalyst or a supported mixed oxide catalyst. The method is based on the precipitation of a single or multi-phase solid by altering the slurry condition, for example, the pH of the solution, applying heat or vaporizing a suitable amount of a precursor, and adsorbing it on a support material. The co-precipitation method slightly differs from the previous one as, in this case, the catalyst is formed by dissolving and mixing the active metal salt and the support to promote nucleation and growth of a combined solid precursor containing both the active element and support.
- (b)
- Impregnation method: In this method, the selected support is immersed in a precursor solution allowing the precursor of the active phase to diffuse into the porous structure of the support. The obtained slurry is slowly dried and later calcined at an appropriate temperature without exceeding the thermal decomposition temperature of the support to prevent its collapse and sinter its texture.
- (c)
- Sol–gel method: This method is very versatile and allows control of the texture, composition, homogeneity, and structural properties of solids, and makes possible production of tailored materials such as dispersed metals, oxidic catalysts, and chemically modified supports. This method involves the formation of a sol from dispersed colloid solutions or from some inorganic precursors as a starting material followed by the formation of a gel. This method yields various configurations such as monoliths, coatings, foams, and fibers without using highly cost processing technologies. The method is based on the hydrolysis and condensation of metal alkoxides such as SiCl4 with alcohol [36,37,38].
- (d)
- Chemical deposition: This method consists of the formation of thin films on a heated substrate via a chemical reaction of gas-phase precursors, which is known as the Chemical Vapor Deposition (CVD) method. A typical example of a catalyst prepared by this method is a 2D transition metal dichalcogenides deposited on thin polymeric films [39,40].
2.4. Characterization Techniques
2.4.1. X-ray Diffraction (XRD)
2.4.2. X-ray Photoelectron Spectroscopy (XPS) or Electron Spectroscopy for Chemical Analysis (ESCA)
2.4.3. Ion Scattering Spectroscopy (ISS)
2.4.4. Electron Microscopy (EM)
- (a)
- Backscattered electrons (BSE): These are high-energy electrons that are ejected from the solid, losing only a small amount of energy. They originate from deep layers on the surface (a few microns deep). They provide information about the composition of the surface and lower-resolution images. These electrons are reflected after elastic interactions between the beam and the sample.
- (b)
- Secondary Electrons (SE): These electrons originate from a few nanometers into the sample surface, with a lower energy compared to the backscattered electrons. They are very sensitive to surface structure and provide topographic information. Secondary electrons are a result of inelastic interactions between the electron beam and the sample.
- (c)
- X-rays: These characteristic X-rays are produced when electrons hit the sample surface. They give information about the elemental composition of the sample.
2.4.5. Infrared Spectroscopy (IR)
2.4.6. Mössbauer Spectroscopy
2.4.7. Thermal Analysis
2.4.8. Adsorption
- (a)
- Hysteresis: Hysteresis is defined as the void space between the adsorption and the desorption branch of the isotherms corresponding to the same solid and is associated with the capillary condensation that occurs when the relative pressure over the sample exceeds 0.4, approximately. The hysteresis is related to the shape of a pore in the solid and occurs when a greater pressure change is required to remove an adsorbate from a pore than what was required to adsorb.
- (b)
- (c)
- t-plot analysis: According to Lippens and de Boer [70,71], a comparison of the adsorption isotherm produced by a solid with a standard isotherm (produced by a non-porous material but with a similar nature of the solid in question) yields information about the micropore volume as well as the external surface area of the solid. The external surface area corresponds to the area in the solid that extends above the micropore area. According to Halsey, one of the t-equations, the t value is evaluated from the inverse of the relative pressure according to Equation (3).
- (d)
- Pore size and structure: This term is related to mesoporous materials and is directly connected with type IV of adsorption isotherm [71], which shows a hysteresis between the adsorption and the desorption branch. According to Kelvin, there is a direct relationship between the size of the pore and the relative pressure in equilibrium withdrawn directly from the adsorption isotherm (Equation (4)). Finally, the pore size would be the sum of the Kelvin radius and the value of the thickness (t) according to the Pierce method (see Equation (5)) [72,73,74].
- (e)
- Total pore volume: The total pore volume of a solid corresponds to the total adsorbed volume at a relative pressure close to unity on the adsorption isotherm, it is normally converted into volume of liquid according to the Gurvitsch rule (see Equation (6)) [75].
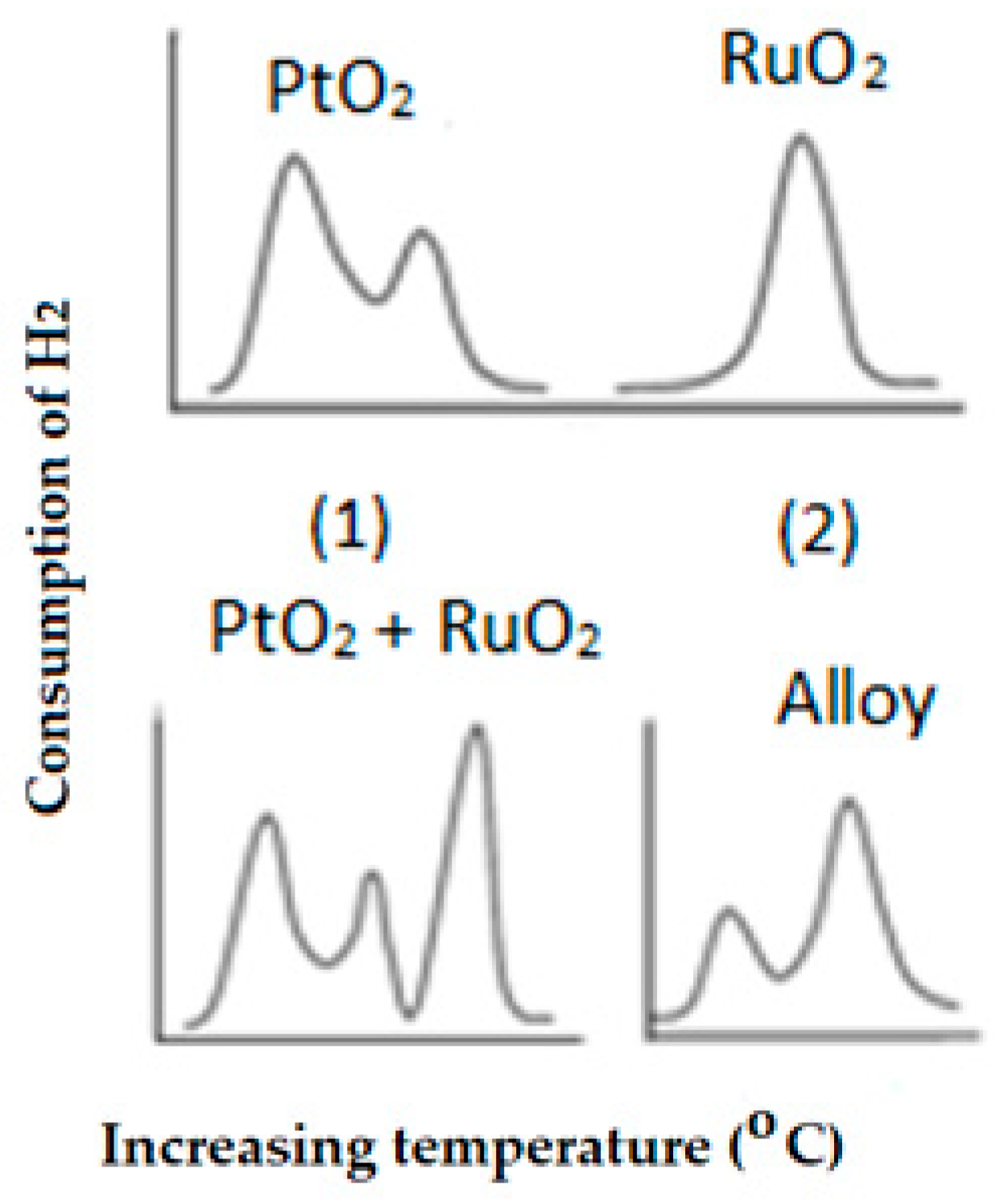
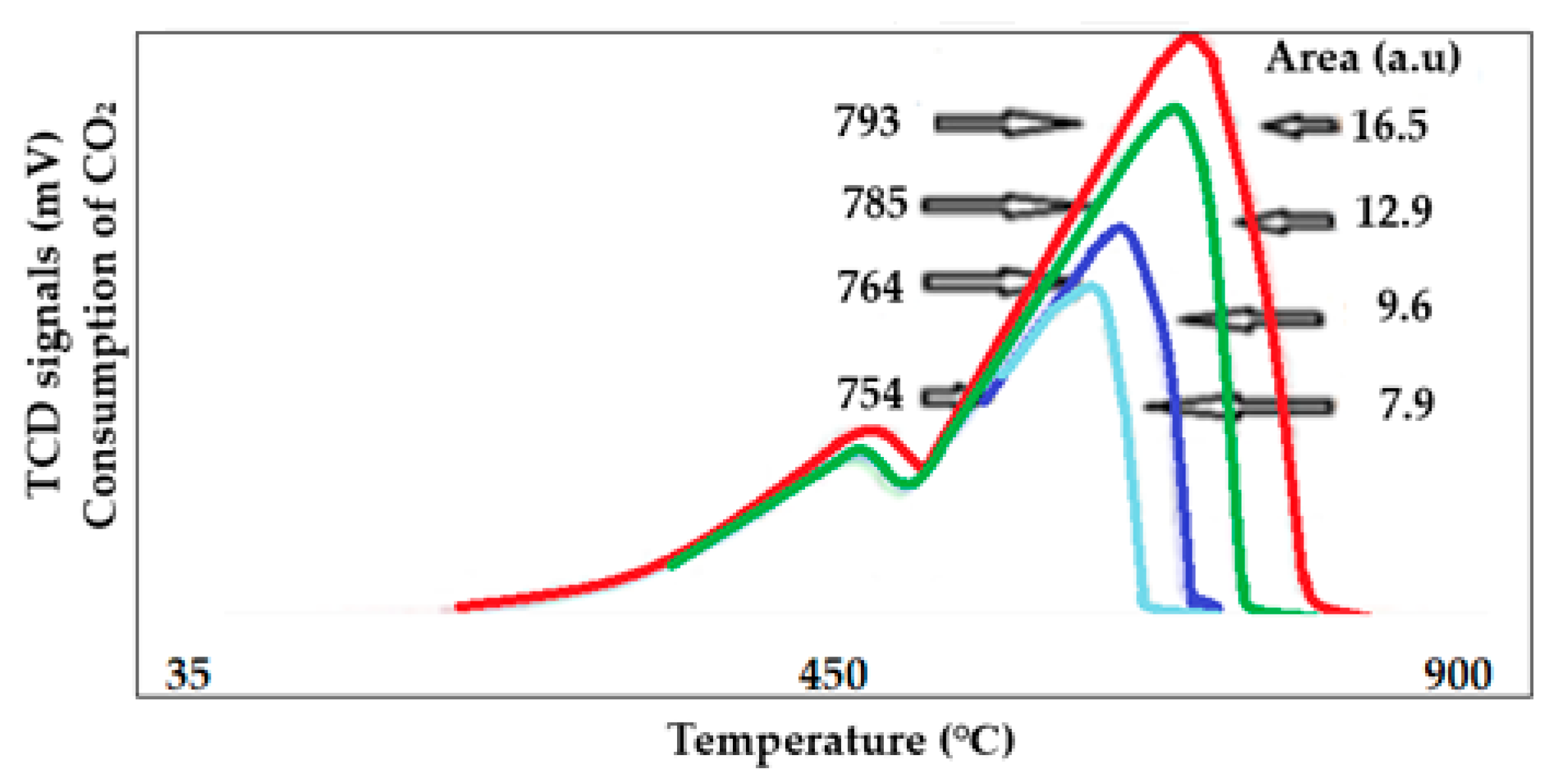
2.4.9. Temperature Programmed Reduction (TPR)
- (a)
- The quantity of the total amount of oxides in the solid;
- (b)
- The maximum reduction temperature of the oxides;
- (c)
- The presence of various particle sizes of the oxide that would be indicated by shoulders on the TPR profile;
- (d)
- Shift on the maximum reduction peak indicating the interaction of the active particles with the support. In other words, the role of the support is to stabilize the active species by a certain type of interaction that could be weak or strong; the stronger that interaction is the higher the reduction temperature.
2.4.10. Temperature Programmed Oxidation (TPO)
2.4.11. Temperature Programmed Reaction (TPRe)
2.4.12. Evaluation of Acid Sites in Solids
2.4.13. Mercury Intrusion
3. Synthesis, Characterization and Catalytic Application
3.1. A Comprehensive Assessment of Characterization Techniques
3.1.1. Physical Adsorption
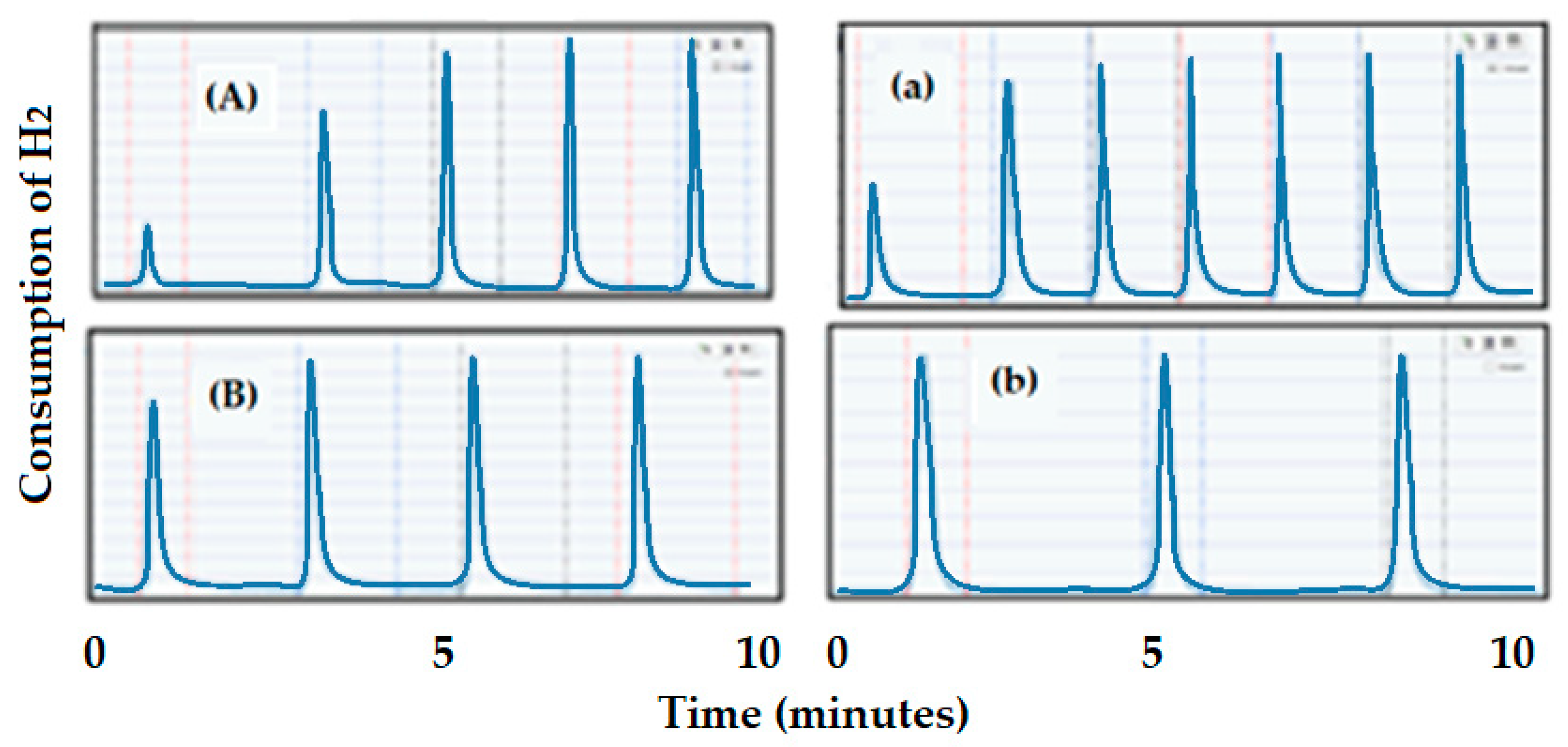
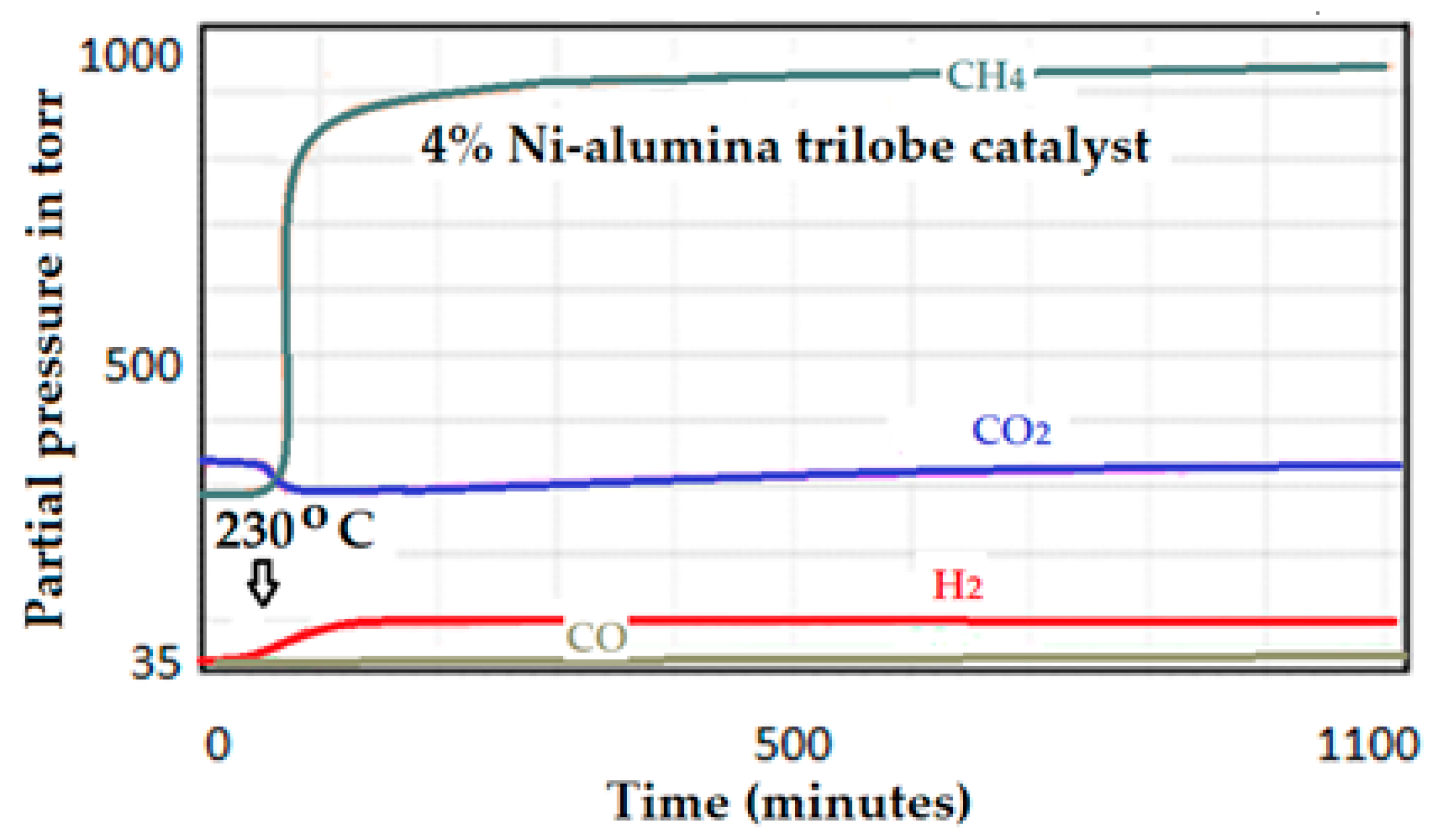
3.1.2. Chemical Adsorption, Chemisorption or Selective Adsorption Technique
- (1)
- The intensity difference of CH4 at the end of the reaction minus the intensity at the start (IT);
- (2)
- Determination of the total amount of the metal on the catalyst (MT);
- (3)
- Determination of the amount of the metal on the surface (MS): multiplying the total amount of metal (MT) by the % dispersion of the metal on the catalyst;
- (4)
- Estimation of the performance obtained by dividing the resulting intensity of the expected product (IT)/MS.
3.1.3. Characterization of Catalysts upon Deactivation
4. Summary, Conclusions, and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bradford, M.C.J.; Vannice, M.A. CO2 reforming of CH4. Catal. Rev. Sci. Eng. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 hydrogentaion to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 2013, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Jalama, K. Carbon dioxide hydrogenation over nickel-, ruthenium-, and copper-based catalysts: Review of kinetics and mechanism. Catal. Rev. Sci. Eng. 2017, 59, 95–164. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Heterogeneous catalysts for hydrogenation of CO2 and bicarbonates to formic acid and formates. Catal. Rev. Sci. Eng. 2018, 60, 566–593. [Google Scholar] [CrossRef]
- Akhundi, A.; Habibi-Yangjeh, A.; Abitorabi, M.; Rahim-Pouran, S. Review on photocatalytic conversion of carbon dioxide to value-added compounds and renewable fuels by graphitic carbon nitride-based photocatalysts. Catal. Rev. Sci. Eng. 2019, 61, 595–628. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogenous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Saad, D.M.; Bilbeisi, R.A.; Alnouri, S.Y. Optimizing network pathways of CO2 conversion processes. J. CO2 Util. 2021, 45, 101433. [Google Scholar] [CrossRef]
- Saini, S.; Prajapati, P.K.; Jain, S.L. Transition metal-catalyzed carboxylation of olefins with Carbon dioxide: A comprehensive review. Catal. Rev. Sci. Eng. 2022, 64, 631–677. [Google Scholar] [CrossRef]
- Latsiou, A.I.; Charisiou, N.D.; Frontistis, Z.; Bansode, A.; Goula, M.A. CO2 hydrogenation for the production of higher alcohols: Trends in catalyst developments, Challenges and opportunities. Catal. Today 2023, 420, 114179. [Google Scholar] [CrossRef]
- Sancho-Sanz, I.; Korili, S.A.; Gil, A. Catalytic valorization of CO2 by hydrogenation: Current status and future trends. Catal. Rev. Sci. Eng. 2023, 65, 698–772. [Google Scholar] [CrossRef]
- Sibi, M.G.; Verma, D.; Kim, J. Direct conversion of CO2 into aromatics over multifunctional heterogeneous catalysts. Catal. Rev. Sci. Eng. 2022; in press. [Google Scholar]
- Xie, S.; Li, Z.; Li, H.; Fang, Y. Integration of carbon capture with heterogeneous catalysis toward methanol production: Chemistry, challenges, and opportunities. Catal. Rev. Sci. Eng. 2023; in press. [Google Scholar] [CrossRef]
- Torrez-Herrera, J.J.; Korili, S.A.; Gil, A. Recent progress in the application of Ni-based catalysts for the dry reforming of methane. Catal. Rev. Sci. Eng. 2021; in press. [Google Scholar]
- Sahu, A.K.; Zhao, X.S.; Upadhyayula, S. Ceria-based photocatalysts in water-splitting for hydrogen production and carbon dioxide reduction. Catal. Rev. Sci. Eng. 2023; in press. [Google Scholar]
- Komiyama, M. Design and preparation of impregnated catalysts. Catal. Rev. Sci. Eng. 2006, 27, 341–372. [Google Scholar] [CrossRef]
- Bourikas, K.; Kordulis, C.; Lycourghiotis, A. The role of the liquid-solid interface in the preparation of supported catalysts. Catal. Rev. Sci. Eng. 2007, 48, 363–444. [Google Scholar] [CrossRef]
- Jeffry, L.; Ong, M.Y.; Nomanbhay, S.; Mofijur, M.; Mubashir, M.; Show, P.L. Greenhouse gases utilization: A review. Fuel 2021, 301, 121017. [Google Scholar] [CrossRef]
- Santamaría, L.; Korili, S.A.; Gil, A. Layered double hydroxides for CO2 adsorption at moderate temperatures: Synthesis and amelioration strategies. Chem. Eng. J. 2023, 455, 140551. [Google Scholar] [CrossRef]
- Mustafa, A.; Lougou, B.G.; Shuai, Y.; Wang, Z.; Tan, H. Current technology development for CO2 utilization into solar fuels and chemicals. A review. J. Energy Chem. 2020, 49, 96–123. [Google Scholar] [CrossRef]
- Rahman, F.A.; Aziz, M.M.A.; Saidur, R.; Abu Bakar, W.A.W.; Hainin, M.R.; Putrajaya, R.; Hassan, N.A. Pollution to solution: Capture and sequestration of carbon dioxide (CO2) and its utilization as a renewable energy source for a sustainable future. Renew. Sustain. Energy Rev. 2017, 71, 112–126. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Dong, F. Grand challenges for catalytic remediation in environmental and energy applications toward a cleaner and sustainable future. Front. Environ. Chem. 2020, 1, 5. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Fazlollahi, F.; Nikoo, M.K.; Sefidkon, F.; Klemeš, J.J.; Baxter, L.L. Mechanisms and kinetics of CO2 hydrogenation to value-added products: A detailed review on current status and future trends. Renew. Sustain. Energy Rev. 2017, 80, 1292–1311. [Google Scholar] [CrossRef]
- Mucsi, Z.; Chass, G.A.; Viskolcz, B.; Csizmadia, I.G. Quantitative scale for the extent of conjugation of carbonyl groups: “Carbonylicity” percentage as a chemical driving force. J. Phys. Chem. A 2008, 112, 9153–9165. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Zhang, J.; Luque, R.; Yan, N. Ni-based bimetallic heterogeneous catalysts for energy and environmental applications. Energy Environ. Sci. 2016, 9, 3314–3347. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential role of the support for nickel-based CO2 methanation catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Le, T.A.; Kang, J.K.; Park, E.D. CO and CO2 methanation over Ni/SiC and Ni/SiO2 catalysts. Top. Catal. 2018, 61, 1537–1544. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Mihet, M.; Dan, M.; Barbu-Tudoran, L.; Lazar, M.D. CO2 Methanation using multimodal Ni/SiO2 catalysts: Effect of support modification by MgO, CeO2, and La2O3. Catalysts 2021, 11, 443. [Google Scholar] [CrossRef]
- Rahat, J.; Tetsuya, N. Effect of texture and physical properties of catalysts on ammonia synthesis. Catal. Today 2022, 397, 592–597. [Google Scholar]
- Rahat, J.; Tetsuya, N. Effect of preparation method and reaction parameters on catalytic activity for ammonia synthesis. Int. J. Hydrogen Energy 2021, 46, 35209–35218. [Google Scholar]
- Cauqui, M.A.; Rodriguez-Izquierdo, J.M. Application of the sol-gel methods to catalyst preparation. J. Non-Cryst. Solids 1992, 147–148, 724–738. [Google Scholar] [CrossRef]
- Pinna, F. Supported metal catalysts preparation. Catal. Today 1998, 41, 129–137. [Google Scholar] [CrossRef]
- Campanati, M.; Fornasari, G.; Vaccari, A. Fundamentals in the preparation of heterogeneous catalysts. Catal. Today 2003, 77, 299–314. [Google Scholar] [CrossRef]
- Serp, P.; Kalck, P.; Feurer, R. Chemical vapor deposition methods for the controlled preparation of supported catalytic materials. Chem. Rev. 2002, 192, 3085–3128. [Google Scholar] [CrossRef]
- Kumar, V.A.; Nagaraja, B.M.; Shashikala, V.; Padmasri, A.H.; Madhavendra, S.S.; Raju, B.D.; Rao, K.S.R. Highly efficient Ag/C catalyst prepared by electro-chemical deposition method in controlling microorganisms in water. J. Mol. Catal. A Chem. 2004, 223, 313–319. [Google Scholar] [CrossRef]
- Gil, A.; García, A.M.; Fernández, M.; Vicente, M.A.; González-Rodríguez, B.; Rives, V.; Korili, S.A. Effect of dopants on the structure of titanium oxide used as a photocatalyst for the removal of emergent contaminants. J. Ind. Eng. Chem. 2017, 53, 183–191. [Google Scholar] [CrossRef]
- Siegbahn, K. Electron spectroscopy for atoms, molecules and condensed matter—An overview. J. Electron Spectrosc. Relat. Phenom. 1985, 36, 113–129. [Google Scholar] [CrossRef]
- Amiri, M.J.; Roohi, R.; Gil, A. Numerical simulation of Cd(II) removal by ostrich bone ash supported nanoscale zero-valent iron in a fixed-bed column system: Utilization of unsteady advection-dispersion-adsorption equation. J. Water Process. Eng. 2018, 25, 1–14. [Google Scholar] [CrossRef]
- Niehus, H.; Bauer, E. Quantitative aspects of ion scattering spectroscopy (ISS). Surf. Sci. 1975, 47, 222–233. [Google Scholar] [CrossRef]
- Aono, M.; Souda, R.; Oshima, C.; Ishizawa, Y. Structure analysis of the Si(111)7 × 7 surface by low-energy ion scattering. Phys. Rev. Lett. 1983, 50, 1293. [Google Scholar] [CrossRef]
- Soares, G.V.; Bastos, K.P.; Pezzi, R.P.; Miotti, L.; Driemeier, C.; Baumvol, I.J.R.; Hinkle, C.; Lucovsky, G. Nitrogen bonding, stability, and transport in AlON films on Si. Appl. Phys. Lett. 2004, 84, 4992–4994. [Google Scholar] [CrossRef]
- Egerton, R.F. An Introduction to TEM, SEM, and AEM. In Physical Principles of Electron Microscopy; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gil, A.; Fernández, M.; Mendizábal, I.; Korili, S.A.; Soto-Armañanzas, J.; Crespo-Durante, A.; Gómez-Polo, C. Fabrication of TiO2 coated metallic wires by the sol–gel technique as a humidity sensor. Ceram. Int. 2016, 42, 9292–9298. [Google Scholar] [CrossRef]
- Cardona, Y.; Korili, S.A.; Gil, A. A nonconventional aluminum source in the production of alumina-pillared clays for the removal of organic pollutants by adsorption. Chem. Eng. J. 2021, 425, 130708. [Google Scholar] [CrossRef]
- Yanyan, C.; Caineng, Z.; Mastalerz, M.; Suyun, H.; Gasaway, C.; Xiaowan, T. Applications of micro-fourier transform infrared spectroscopy (FTIR) in the geological sciences-A review. Int. J. Mol. Sci. 2015, 16, 30223–30250. [Google Scholar]
- De la Puente, G.; Centeno, A.; Gil, A.; Grange, P. Interactions between molybdenum and activated carbons on the preparation of activated carbon-supported molybdenum catalysts. J. Colloid Interface Sci. 1998, 202, 155–166. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Gibb, T.C. Mössbauer Spectroscopy; University of Leeds: Leeds, UK, 1971. [Google Scholar]
- Gil, A.; Korili, S.A.; Trujillano, R.; Vicente, M.A. A review on characterization of pillared clays by specific techniques. Appl. Clay Sci. 2011, 53, 97–105. [Google Scholar] [CrossRef]
- Moura, J.A.; Araujo, A.S.; Coutinho, A.C.S.L.S.; Aquino, J.M.F.B.; Silva, A.O.S.; Souza, M.J.B. Thermal analysis applied to characterization of copper and nickel catalysts. J. Therm. Anal. Calorim. 2005, 79, 435–438. [Google Scholar] [CrossRef]
- Tursunov, O.; Zubek, K.; Czerski, G.; Dobrowolski, J. Studies of CO2 gasification of the Miscanthus giganteus biomass over Ni/Al2O3-SiO2 and Ni/Al2O3-SiO2 with K2O promoter as catalysts. J. Therm. Anal. Calorim. 2020, 139, 3481–3492. [Google Scholar] [CrossRef]
- Jiménez, A.; Misol, A.; Morato, A.; Rives, V.; Vicente, M.A.; Gil, A. Optimization of hydrocalumite preparation under microwave irradiation for recovering aluminium from a saline slag. Appl. Clay Sci. 2021, 212, 106217. [Google Scholar] [CrossRef]
- Vizcaíno, A.J.; Carrero, A.; Calles, J.A. Hydrogen production by ethanol steam reforming over Cu-Ni supported catalysts. Int. J. Hydrogen Energy 2007, 32, 1450–1461. [Google Scholar] [CrossRef]
- Jiménez, A.; Misol, A.; Morato, A.; Rives, V.; Vicente, M.A.; Gil, A. Synthesis of pollucite and analcime zeolites by recovering aluminum from a saline slag. J. Clean. Prod. 2021, 297, 126667. [Google Scholar] [CrossRef]
- Webb, P.A.; Orr, C. Analytical Method in Fine Particle Technology; Micromeritics Instrument Corporation: Norcross, GA, USA, 1995. [Google Scholar]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity, 2nd ed.; Academic Press: Cambridge, MA, USA, 1982. [Google Scholar]
- Gil, A. Análisis Textural de Solidos Porosos Mediante Adsorción Física de Gases; Universidad Pública de Navarra: Pamplona, Spain, 2020. [Google Scholar]
- Bond, G.C. Heterogeneous Catalysts: Principles and Applications; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Baiker, A. Experimental methods for the characterization of catalysts. I. Gas adsorption methods, pycnometry and porosimetry. Int. Chem. Eng. 1985, 25, 16. [Google Scholar]
- Delannay, F. Characterization of Heterogeneous Catalysts; Marcel Dekker: New York, NY, USA, 1984. [Google Scholar]
- Yunes, S. Characterization and Catalytic Tests for Laterite and Bauxite. Ph.D. Thesis, Université Catholique de Louvain, Louvain-la-Neuce, Belgium, 1984. [Google Scholar]
- Somorjai, G.A. Introduction to Surface Chemistry and Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1994. [Google Scholar]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. Part I. Solids and liquids. J. Am. Chem. Soc. 1916, 38, 2221–2295. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lippens, B.C.; de Boer, J.H. Studies on pore systems in catalysts: V. The t method. J. Catal. 1965, 4, 319–323. [Google Scholar] [CrossRef]
- Lippens, B.C.; de Boer, J.H. Studies on pore systems in catalysts I. The adsorption of nitrogen; apparatus and calculation. J. Catal. 1964, 3, 32–37. [Google Scholar] [CrossRef]
- Pierce, C.; Ewings, B. Areas of uniform graphite surfaces. J. Phys. Chem. 1964, 68, 2562–2568. [Google Scholar] [CrossRef]
- Pierce, C. Localized adsorption on graphite and absolute surface areas. J. Phys. Chem. 1969, 73, 813–817. [Google Scholar] [CrossRef]
- Davis, B.W.; Varsanik, R.G. A study of crystalline titanium carbide. J. Colloid Interface Sci. 1971, 37, 870–878. [Google Scholar] [CrossRef]
- Gurvitsch, L. Physicochemical attractive force. J. Phys. Chem. Soc. Russ. 1915, 47, 805–827. [Google Scholar]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Hofmann, A.W. Researches into the molecular constitution of the organic bases. Philos. Trans. R. Soc. Lond. 1851, 141, 357–398. [Google Scholar]
- Hofmann, A.W. Übergang der flüchtigen basen in eine reihe nichtflüchtige alkaloide. Ann. Chem. Pharms. 1851, 78, 253–286. [Google Scholar]
- Hofmann, A.W. Continuation of Übergang der flüchtigen basen in eine reihe nichtflüchtige alkaloide. Ann. Chem. Pharms. 1851, 79, 11–39. [Google Scholar] [CrossRef]
- Wachs, I.E. Number of surface sites and turnover frequencies for oxide catalysts. J. Catal. 2022, 405, 462–472. [Google Scholar] [CrossRef]
- Washburn, E.W. Note on a method of determining the distribution of pore sizes in a porous material. Proc. Natl. Acad. Sci. USA 1921, 7, 115–116. [Google Scholar] [CrossRef]
- Gil, A. Classical and new insights into the methodology for characterizing adsorbents and metal catalysts by chemical adsorption. Catal. Today 2023, 423, 114016. [Google Scholar] [CrossRef]
- Sabatier, P.; Senderens, J. Direct hydrogenation of oxides of carbon in presence of various finely divided metals. CR Acad Sci. 1902, 134, 689–691. [Google Scholar]

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2/g) | VpT (cm3(STP)/g) | Pore Diameter (nm) |
|---|---|---|---|
| Alumina Trilobe | 310 | 0.67 | 8.6 |
| Alumina pellet | 92 | 0.26 | 10.9 |
| 13 wt.% CuO-alumina catalyst | 165 | 0.38 | 9.1 |
| Catalyst | SBET (m2/g) | VpT (cm3(STP)/g) | Pore Diameter (nm) |
|---|---|---|---|
| 14.5 wt.% Ni (A) | 222 | 0.51 | 8.9 |
| 4% wt.Ni (B) | 280 | 0.53 | 8.7 |
| 11% wt.Ni (C) | 75 | 0.19 | 10.0 |
| 5.6% wt.Ni (D) | 83 | 0.21 | 10.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yunes, S.; Kenvin, J.; Gil, A. On the Genesis of a Catalyst: A Brief Review with an Experimental Case Study. Eng 2023, 4, 2375-2406. https://doi.org/10.3390/eng4030136
Yunes S, Kenvin J, Gil A. On the Genesis of a Catalyst: A Brief Review with an Experimental Case Study. Eng. 2023; 4(3):2375-2406. https://doi.org/10.3390/eng4030136
Chicago/Turabian StyleYunes, Simón, Jeffrey Kenvin, and Antonio Gil. 2023. "On the Genesis of a Catalyst: A Brief Review with an Experimental Case Study" Eng 4, no. 3: 2375-2406. https://doi.org/10.3390/eng4030136