
Correlation between Blood Monocytes and CSF Tau in Alzheimer’s Disease: The Effect of Gender and Cognitive Decline
 , and
, and
Abstract
:1. Introduction
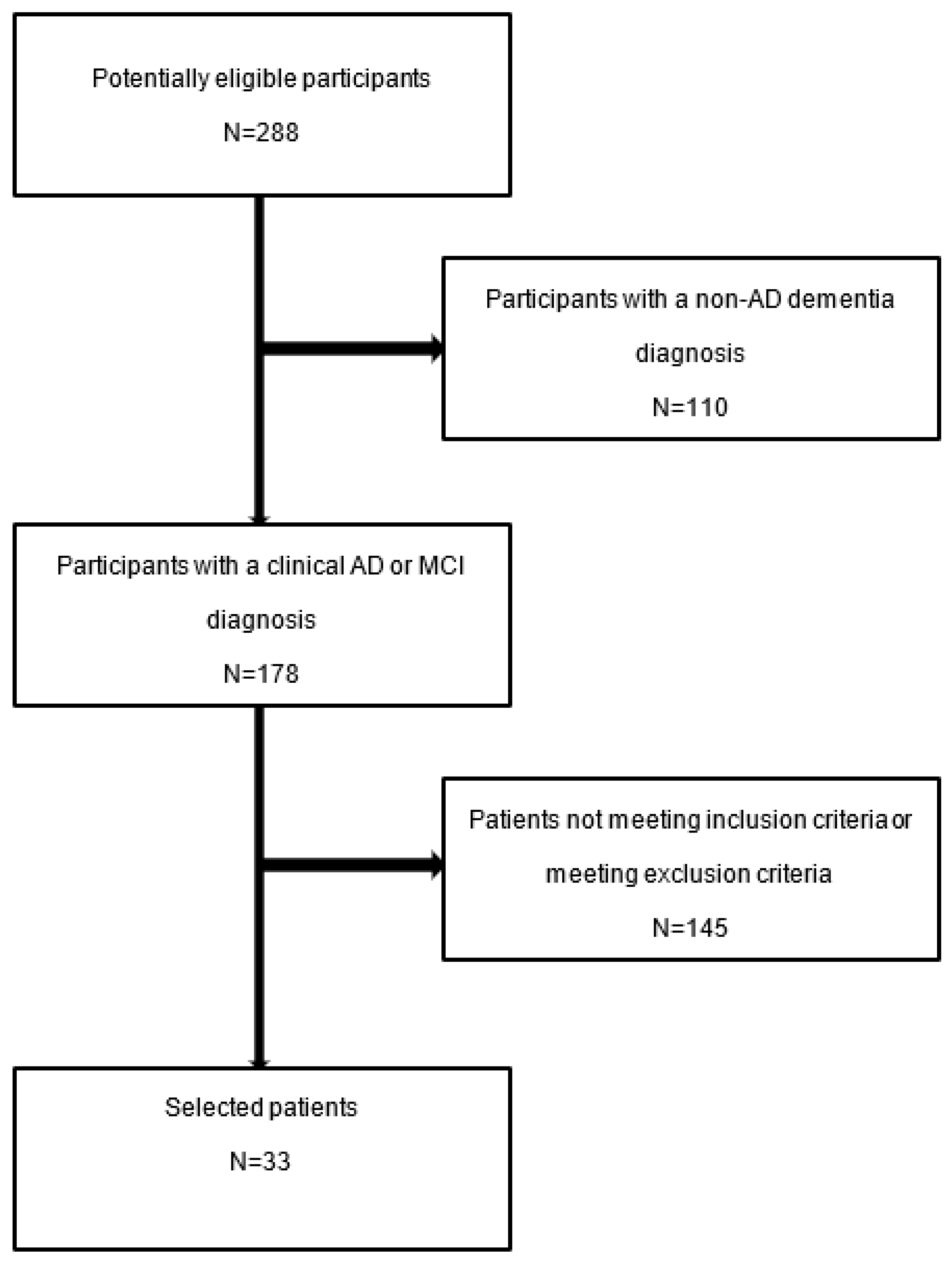
2. Materials and Methods
2.1. Subjects
2.2. Statistical Analyses
3. Results
3.1. Description of the Data
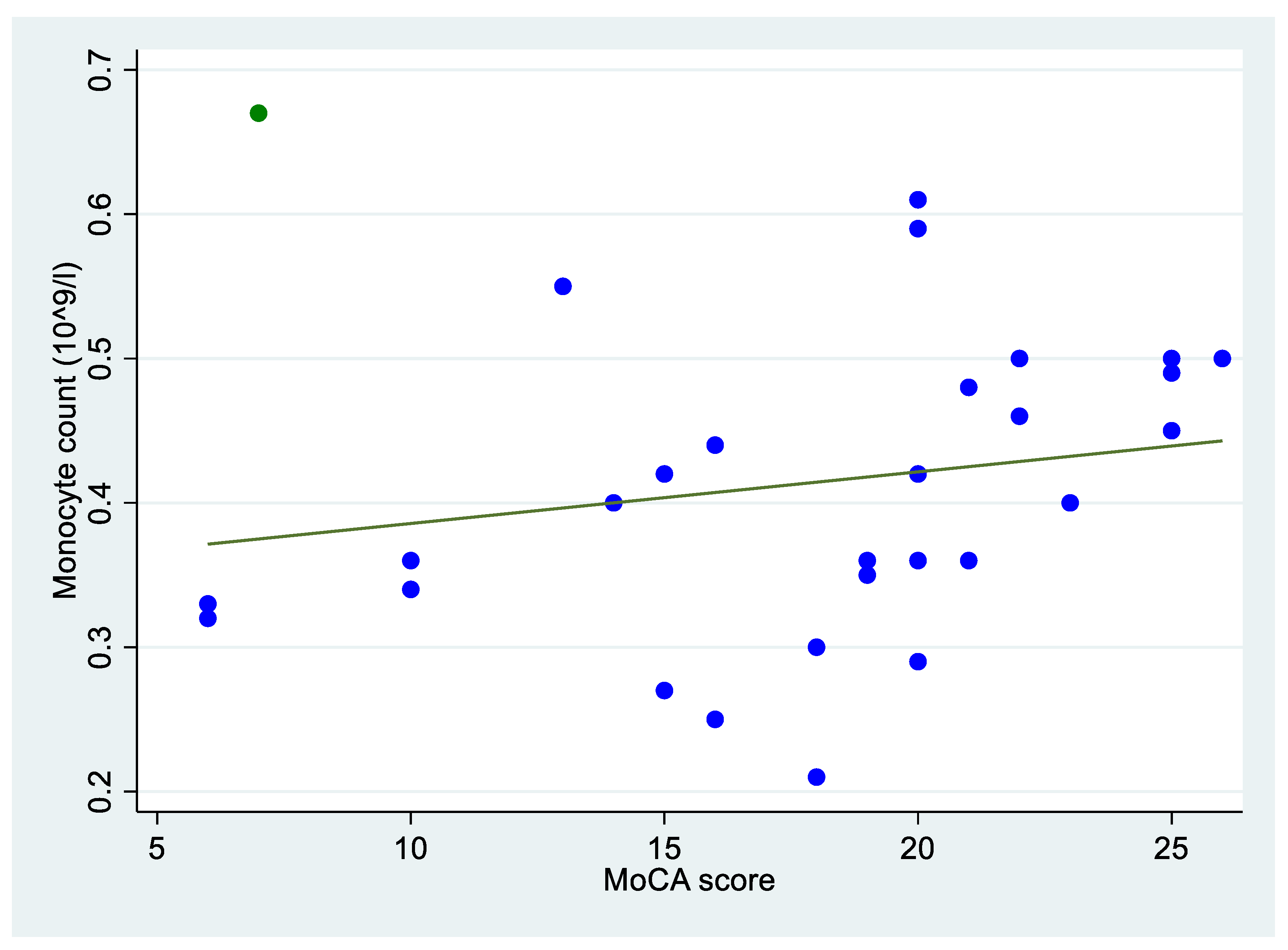
3.2. Bivariate Analysis
4. Discussion
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β protein |
| Aβ42 | Amyloid-β with 42 residues |
| CCR2 | C-C motif chemokine receptor 2 |
| CDR score | Clinical Dementia Rating score |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CX3CR1 | C-X3-C motif chemokine receptor 1 |
| FDG | [18F] Fluorodeoxyglucose |
| GWAS | Genome-wide association studies |
| MCI | Mild cognitive impairment |
| MMSE | Mini mental state examination |
| MoCA | Montreal Cognitive Assessment |
| MRI | Magnetic resonance imaging |
| MS | Multiple sclerosis |
| NFTs | Neurofibrillary tangles |
| NIA-AA | National Institute on Aging and Alzheimer’s Association |
| p-Tau | Phosphorylated tau |
| PET | Positron emission tomography |
| t-Tau | Total tau |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| WHO | World Health Organization |
References
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 15 March 2023).
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epide-miology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Javaid, S.F.; Giebel, C.; Khan, M.A.; Hashim, M.J. Epidemiology of Alzheimer’s disease and other dementias: Rising global burden and forecasted trends. F1000Research 2021, 10, 425. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s Disease: Pathogeny, Etiology, and Related Therapeutic Directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Nal. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, N.; García-Sierra, F.; Wuu, J.; Leurgans, S.; Bennett, D.A.; Berry, R.W.; Binder, L.I. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 2002, 17, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 11, 510–514. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 121–131. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Madhusudan, A.; Doehner, J.; Vogel, P.; Notter, T.; Imhof, C.; Manalastas, A.; Hilfiker, M.; Pfister, S.; Schwerdel, C.; et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation 2012, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Antel, J. Multiple sclerosis-emerging concepts of disease pathogenesis. J. Neuroimmunol. 1999, 98, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Meinl, E.; Weber, F.; Zipp, F.; Schmidt, S.; Sotgiu, S.; Goebels, N.; Voltz, R.; Spuler, S.; Iglesias, A. The role of autoimmune T lymphocytes in the pathogenesis of multiple sclerosi. Neurology 1995, 45, 33–38. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Streit, W.J. Microglia and Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2004, 77, 1–8. [Google Scholar] [CrossRef]
- Butovsky, O.; Kunis, G.; Koronyo-Hamaoui, M.; Schwartz, M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur. J. Neurosci. 2007, 26, 413–416. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Liu, P.T.; Espinosa-Jeffrey, A.; Rosenthal, M.J.; Bernard, G.; Ringman, J.M.; Sayre, J.; Zhang, L.; Zaghi, J.; Dejbakhsh, S.; et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc. Natl. Acad. Sci. USA 2007, 104, 12849–12854. [Google Scholar] [CrossRef]
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. 2005, 7, 221–232; discussion 255–262. [Google Scholar] [CrossRef]
- Saresella, M.; Marventano, I.; Calabrese, E.; Piancone, F.; Rainone, V.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhao, Z.; Zhang, R.; Chen, P.; Zhang, X.; Cheng, F.; Gou, X. Monocytes in the Peripheral Clearance of Amyloid-β and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Daley, D.; Black, K.L.; Koronyo-Hamaoui, M. Clearance of cerebral Aβ in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell. Mol. Life Sci. 2017, 74, 2167–2201. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Zhou, F.Y.; Chen, D.W.; Tan, C.R.; Zeng, G.H.; Liu, Y.H.; Wang, J.; Bu, X.L.; Wang, Y.J.; Li, H.Y.; et al. The Correlation of Tau Levels with Blood Monocyte Count in Patients with Alzheimer’s Disease. Alzheimers Dis. 2022, 85, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 2005, 67, 695–699. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Bergeron, D.; Flynn, K.; Verret, L.; Poulin, S.; Bouchard, R.W.; Bocti, C.; Fülöp, T.; Lacombe, G.; Gauthier, S.; Nasreddine, Z.; et al. Multicenter Validation of an MMSE-MoCA Conversion Table. J. Am. Geriatr. Soc. 2017, 65, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Pinto, T.C.C.; Machado, L.; Bulgacov, T.M.; Rodrigues-Júnior, A.L.; Costa, M.L.G.; Ximenes, R.C.C.; Sougey, E.B. Is the Montreal Cognitive Assessment (MoCA) screening superior to the Mini-Mental State Examination (MMSE) in the detection of mild cognitive impairment (MCI) and Alzheimer’s Disease (AD) in the elderly? Int. Psychogeriatr. 2019, 31, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.M.; Cho, Y.S.; Park, S.; Lee, B.H.; Sohn, B.K.; Choi, C.H.; Choi, J.S.; Jeong, H.Y.; Cho, S.J.; Lee, J.H.; et al. Montreal cognitive assessment reflects cognitive reserve. BMC Geriatr. 2018, 18, 261. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- López-González, I.; Schlüter, A.; Aso, E.; Garcia-Esparcia, P.; Ansoleaga, B.; LLorens, F.; Carmona, M.; Moreno, J.; Fuso, A.; Portero-Otin, M.; et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: Correlations with plaques, tangles, and oligomeric species. J. Neuropathol. Exp. Neurol. 2015, 74, 319–344. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Ko, M.K.; Koronyo, Y.; Azoulay, D.; Seksenyan, A.; Kunis, G.; Pham, M.; Bakhsheshian, J.; Rogeri, P.; Black, K.L.; et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem 2009, 111, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef]
- Majerova, P.; Zilkova, M.; Kazmerova, Z.; Kovac, A.; Paholikova, K.; Kovacech, B.; Zilka, N.; Novak, M. Microglia display modest phagocytic capacity for extracellular tau oligomers. J. Neuroinflammation 2014, 11, 161. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef]
- Vogels, T.; Murgoci, A.N.; Hromádka, T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun. 2019, 7, 109. [Google Scholar] [CrossRef]
- Tang, M.; Harrison, J.; Deaton, C.A.; Johnson, G.V.W. Tau Clearance Mechanisms. Adv. Exp. Med. Biol. 2019, 1184, 57–68. [Google Scholar] [CrossRef]
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of Microglial and Monocyte Chemokines and Their Receptors in Regulating Alzheimer’s Disease-Associated Amyloid-β and Tau Pathologies. Front. Neurol. 2018, 9, 549. [Google Scholar] [CrossRef]
- Xin, S.H.; Tan, L.; Cao, X.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Guedes, J.R.; Santana, I.; Cunha, C.; Duro, D.; Almeida, M.R.; Cardoso, A.M.; de Lima, M.C.; Cardoso, A.L. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement. 2015, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Kim, D.; Katsumoto, A.; Kokiko-Cochran, O.N.; Lamb, B.T.; Ransohoff, R.M. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J. Neuroinflammation 2015, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Lee, D.C.; Hunt, J.B., Jr.; Morganti, J.M.; Selenica, M.L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Wang, J.; Jin, W.S.; Bu, X.L.; Zeng, F.; Huang, Z.L.; Li, W.W.; Shen, L.L.; Zhuang, Z.Q.; Fang, Y.; Sun, B.L.; et al. Physiological clearance of tau in the periphery and its therapeutic potential for tauopathies. Acta Neuropathol. 2018, 136, 525–536. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Prà, I.; Rossi, F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef]
- Gold, M.; El Khoury, J. β-amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin. Immunopathol. 2015, 37, 607–611. [Google Scholar] [CrossRef]
- Stefani, A.; Martorana, A.; Bernardini, S.; Panella, M.; Mercati, F.; Orlacchio, A.; Pierantozzi, M. CSF markers in Alzheimer disease patients are not related to the different degree of cognitive impairment. J. Neurol. Sci. 2006, 251, 124–128. [Google Scholar] [CrossRef]
- Kester, M.I.; van der Vlies, A.E.; Blankenstein, M.A.; Pijnenburg, Y.A.; van Elk, E.J.; Scheltens, P.; van der Flier, W.M. CSF biomarkers predict rate of cognitive decline in Alzheimer disease. Neurology 2009, 73, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Sämgård, K.; Zetterberg, H.; Blennow, K.; Hansson, O.; Minthon, L.; Londos, E. Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int. J. Geriatr. Psychiatry 2010, 25, 403–410. [Google Scholar] [CrossRef] [PubMed]
- van der Vlies, A.E.; Verwey, N.A.; Bouwman, F.H.; Blankenstein, M.A.; Klein, M.; Scheltens, P.; van der Flier, W.M. CSF biomarkers in relationship to cognitive profiles in Alzheimer disease. Neurology 2009, 72, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Lavados, M.; Guillón, M.; Mujica, C.; Bosch, R.; Farías, G.; Fuentes, P. Anomalously phosphorylated tau and Abeta fragments in the CSF correlates with cognitive impairment in MCI subjects. Neurobiol. Aging 2006, 27, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; et al. Sex differences in Alzheimer disease-the gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–469. [Google Scholar] [CrossRef]
- Hua, X.; Hibar, D.P.; Lee, S.; Toga, A.W.; Jack, C.R., Jr.; Weiner, M.W.; Thompson, P.M. Alzheimer’s Disease Neuroimaging Initiative. Sex and age differences in atrophic rates: An ADNI study with n=1368 MRI scans. Neurobiol. Aging 2010, 31, 1463–1480. [Google Scholar] [CrossRef]
- Zhu, D.; Montagne, A.; Zhao, Z. Alzheimer’s pathogenic mechanisms and underlying sex difference. Cell. Mol. Life Sci. 2021, 78, 4907–4920. [Google Scholar] [CrossRef] [PubMed]
- Kodama, L.; Guzman, E.; Etchegaray, J.I.; Li, Y.; Sayed, F.A.; Zhou, L.; Zhou, Y.; Zhan, L.; Le, D.; Udeochu, J.C.; et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat. Neurosci. 2020, 23, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Sayed, F.A.; Kodama, L.; Fan, L.; Carling, G.K.; Udeochu, J.C.; Le, D.; Li, Q.; Zhou, L.; Wong, M.Y.; Horowitz, R.; et al. AD-linked R47H-TREM2 mutation induces disease-enhancing microglial states via AKT hyperactivation. Sci. Transl. Med. 2021, 13, eabe3947. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Yona, S. Inherited and Environmental Factors Influence Human Monocyte Heterogeneity. Front. Immunol. 2019, 10, 2581. [Google Scholar] [CrossRef]
- Wu, K.M.; Zhang, Y.R.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T. The role of the immune system in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101409. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variables | Median (Mean) | Interquartile Range |
|---|---|---|
| Sociodemographics | ||
| Gender (N = 33) | ||
| Men (count) | 15 | - |
| Women (count) | 18 | - |
| Age (N = 33) | 70.00 (70.24) | 13.50 |
| Years of education (N = 33) | 13.00 (12.30) | 4.00 |
| CSF AD biomarkers | ||
| t-Tau [ng/L] (N = 30) | 502.50 | 358.50 |
| p-Tau [ng/L] (N = 29) | 109.00 | 69.50 |
| Aβ42 [ng/L] (N = 30) | 495.50 | 245.00 |
| Aβ40/42 ratio (N = 22) | 0.07 | 0.03 |
| MoCA score (N = 29) | 19.00 (17.66) | 7.00 |
| CDR (N = 33) | 1.00 (1.27) | 1.50 |
| Blood monocyte count [109/L] (N = 33) | 0.40 | 0.17 |
| Variables | Blood Monocyte Count [109/L] | ||
|---|---|---|---|
| Spearman Correlation Coefficients | Mann–Whitney Test | ||
| Median (IQR) | Test Result | ||
| Sociodemographics | |||
| Gender (N = 33) | |||
| Men | - | 0.44 (0.14) | z = −1.739 * |
| Women | - | 0.36 (0.19) | |
| Age (N = 33) | 0.237 | - | |
| CSF AD biomarkers | |||
| t-Tau [ng/L] (N = 30) | −0.368 ** | - | |
| p-Tau [ng/L] (N = 29) | −0.277 | - | |
| Aβ42 [ng/L] (N = 30) | −0.058 | - | |
| Aβ40/42 ratio (N = 22) | 0.091 | - | |
| MoCA score (N = 29) | 0.390 ** | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimiotti, C.G.V.; Paganetti, P.; Rossi, S.; Soldini, E.; Sacco, L. Correlation between Blood Monocytes and CSF Tau in Alzheimer’s Disease: The Effect of Gender and Cognitive Decline. NeuroSci 2023, 4, 319-330. https://doi.org/10.3390/neurosci4040026
Cimiotti CGV, Paganetti P, Rossi S, Soldini E, Sacco L. Correlation between Blood Monocytes and CSF Tau in Alzheimer’s Disease: The Effect of Gender and Cognitive Decline. NeuroSci. 2023; 4(4):319-330. https://doi.org/10.3390/neurosci4040026
Chicago/Turabian StyleCimiotti, Carlotta Ginevra Valentina, Paolo Paganetti, Stefania Rossi, Emiliano Soldini, and Leonardo Sacco. 2023. "Correlation between Blood Monocytes and CSF Tau in Alzheimer’s Disease: The Effect of Gender and Cognitive Decline" NeuroSci 4, no. 4: 319-330. https://doi.org/10.3390/neurosci4040026