
Synthesis and Biological Evaluation of Substituted Fused Dipyranoquinolinones
 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
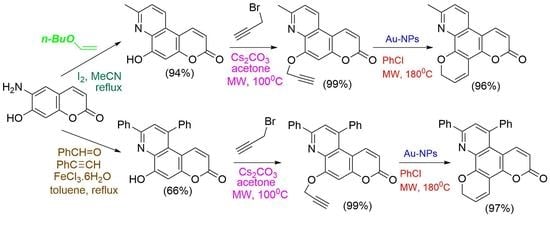
2.1. Chemistry
2.2. Biology
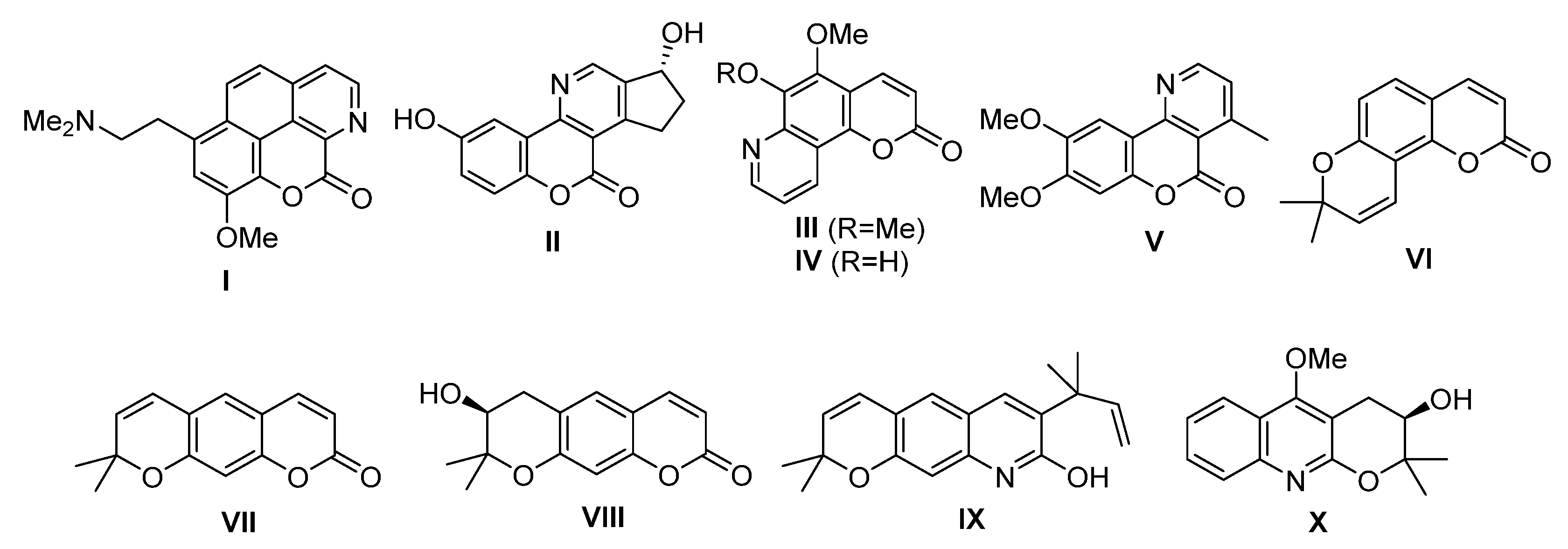
2.3. Docking Studies on Soybean Lipoxygenase
3. Materials and Methods
3.1. Materials
3.2. Chemistry
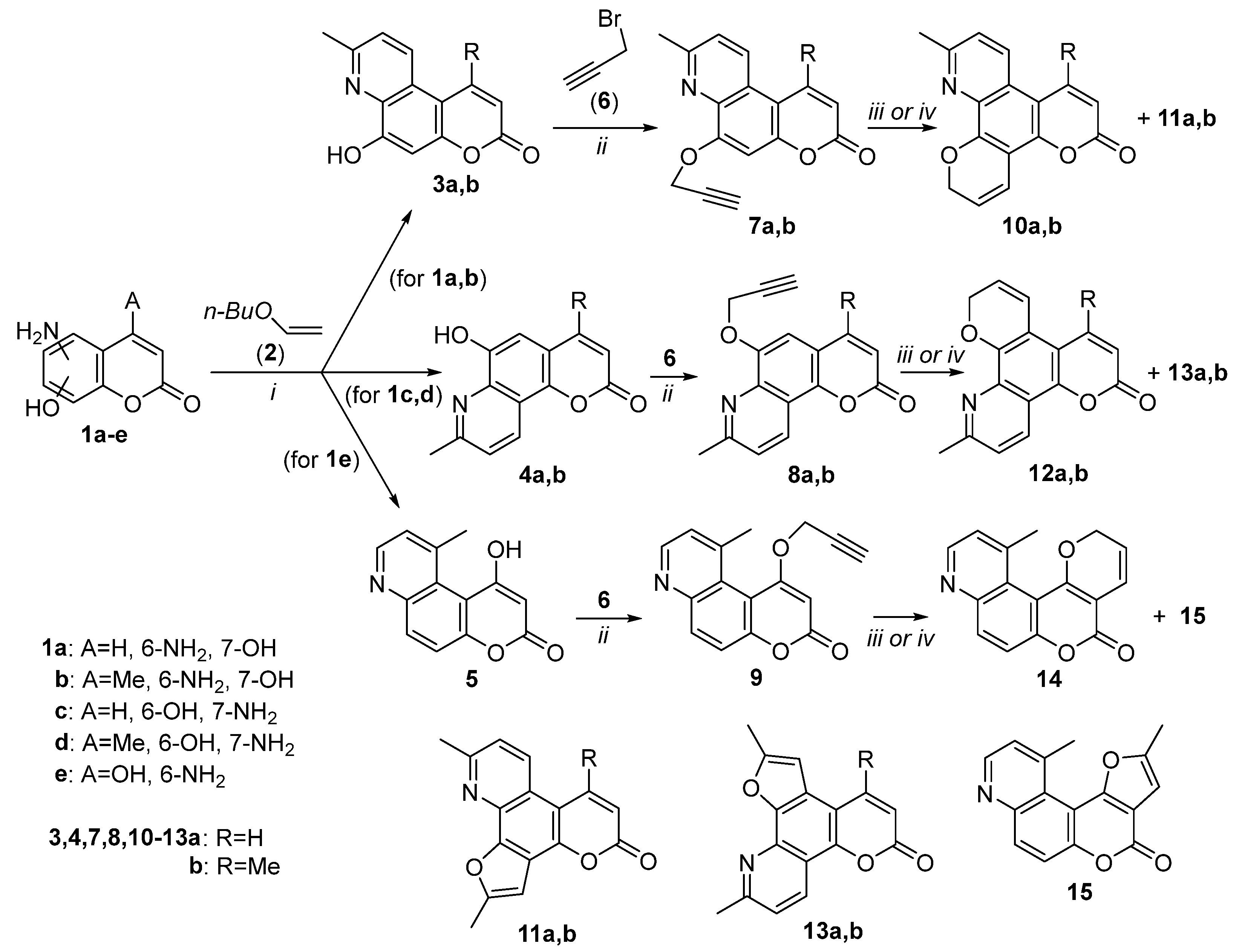
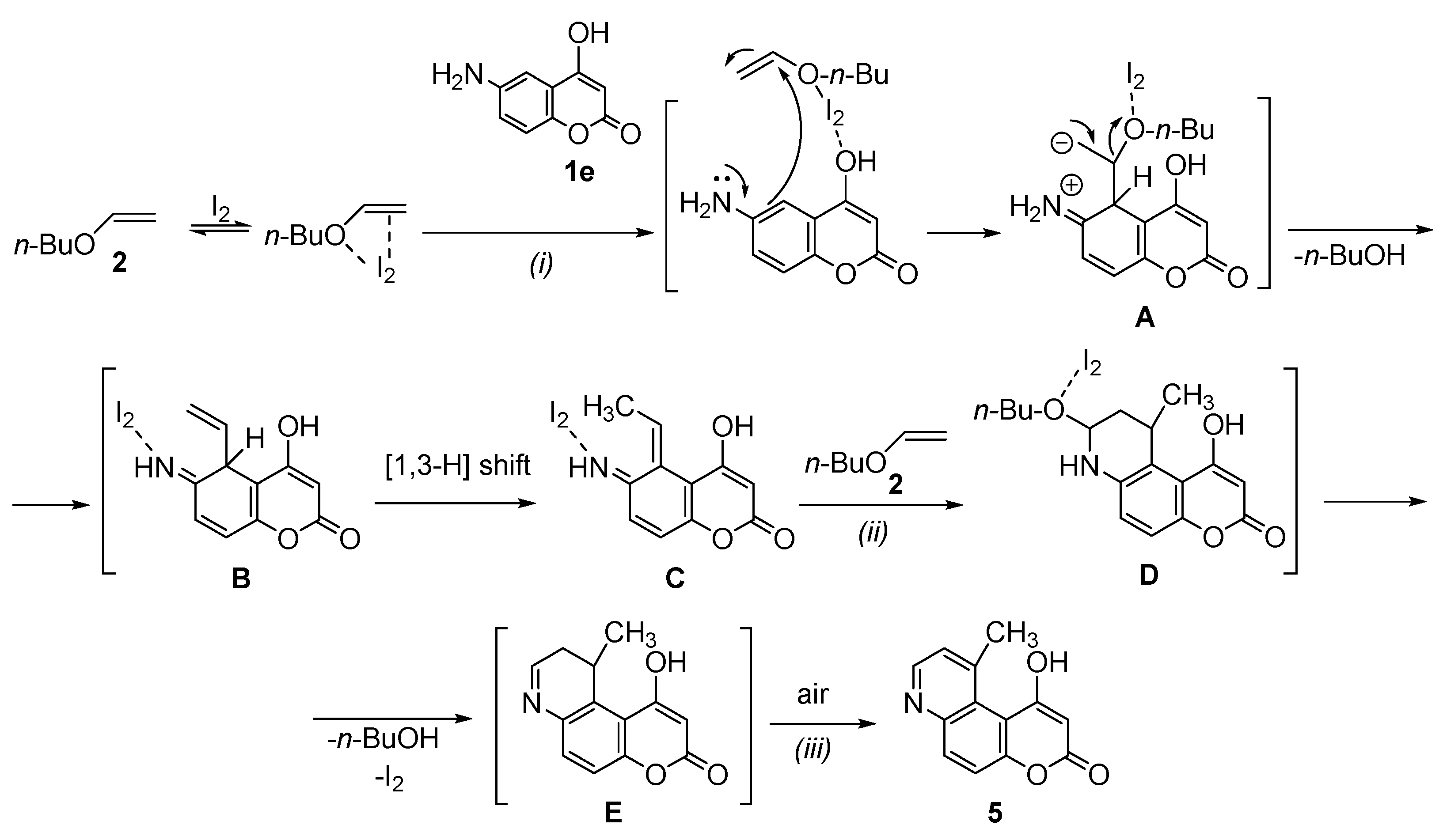
3.2.1. General Procedure for the Synthesis of Methyl-Substituted Pyridine Moiety of Pyranoquinolinones—Synthesis of 6-Hydroxy-8-methyl-3H-pyrano[3,2-f]quinolin-3-one (3a)
3a, Yellow Crystals, m.p. 199–200 °C (Ethyl Acetate)
6-Hydroxy-1,8-dimethyl-3H-pyrano[3,2-f]quinolin-3-one (3b)
6-Hydroxy-8-methyl-2H-pyrano[2,3-f]quinolin-2-one (4a)
6-Hydroxy-4,8-dimethyl-2H-pyrano[2,3-f]quinolin-2-one (4b)
1-Hydroxy-10-methyl-3H-pyrano[3,2-f]quinolin-3-one (5)
3.2.2. General Procedure for the Propargylation of Hydroxypyranoquinolinones—Synthesis of 8-Methyl-6-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (7a)
7a, Light Yellow Crystals, m.p. 121–123 °C (DCM/Hexane)
1,8-Dimethyl-6-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (7b)
8-Methyl-6-(prop-2-yn-yloxy)-2H-pyrano[2,3-f]quinolin-2-one (8a)
4,8-Dimethyl-6-(prop-2-yn-1-yloxy)-2H-pyrano[2,3-f]quinolin-2-one (8b)
10-Methyl-1-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (9)
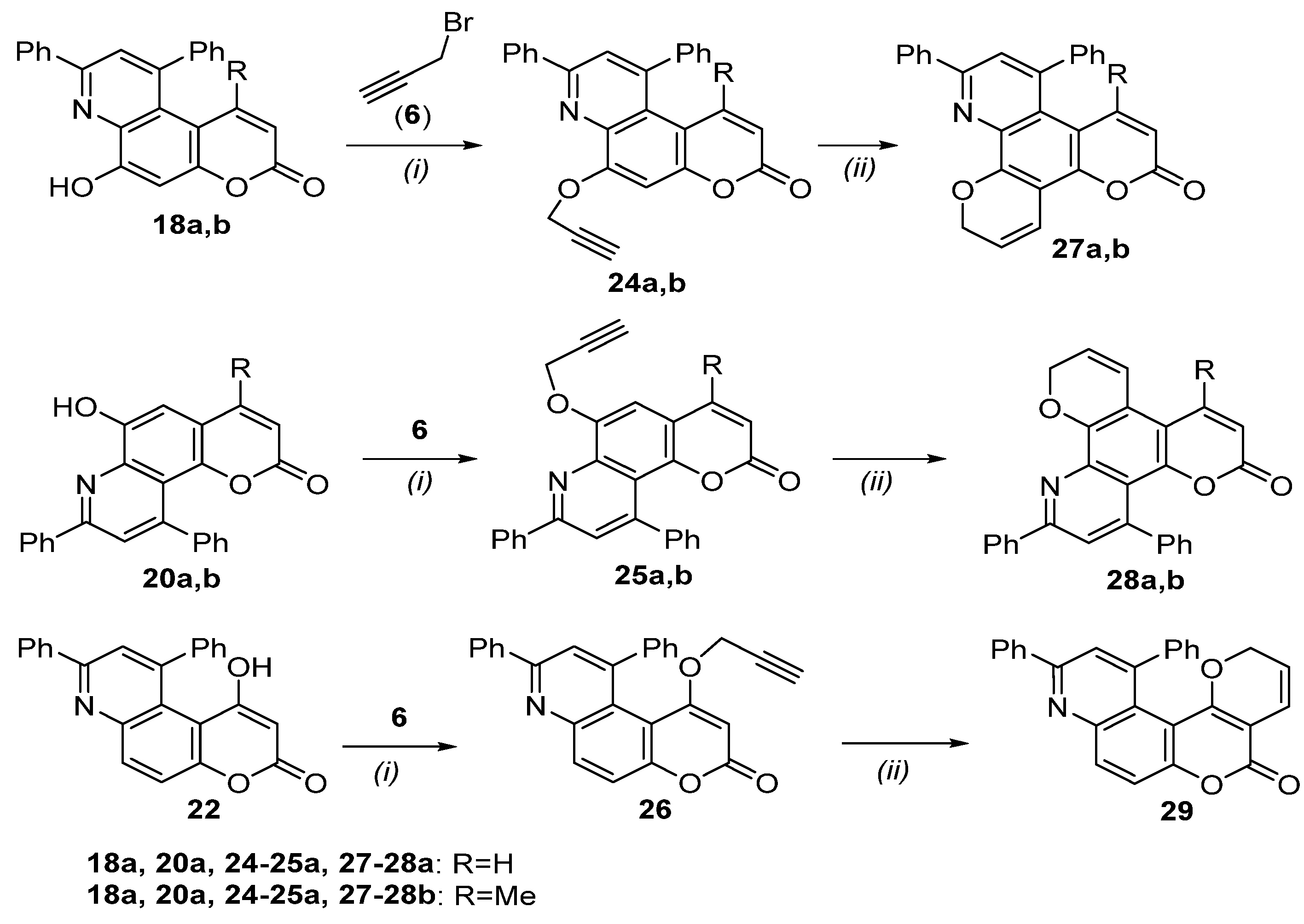
8,10-Diphenyl-6-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (24a)
1-Methyl-8,10-diphenyl-6-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (24b)
8,10-Diphenyl-6-(prop-2-yn-1-yloxy)-2H-pyrano[2,3-f]quinolin-2-one (25a)
4-Methyl-8,10-diphenyl-6-(prop-2-yn-1-yloxy)-2H-pyrano[2,3-f]quinolin-2-one (25b)
8,10-Diphenyl-1-(prop-2-yn-1-yloxy)-3H-pyrano[3,2-f]quinolin-3-one (26)
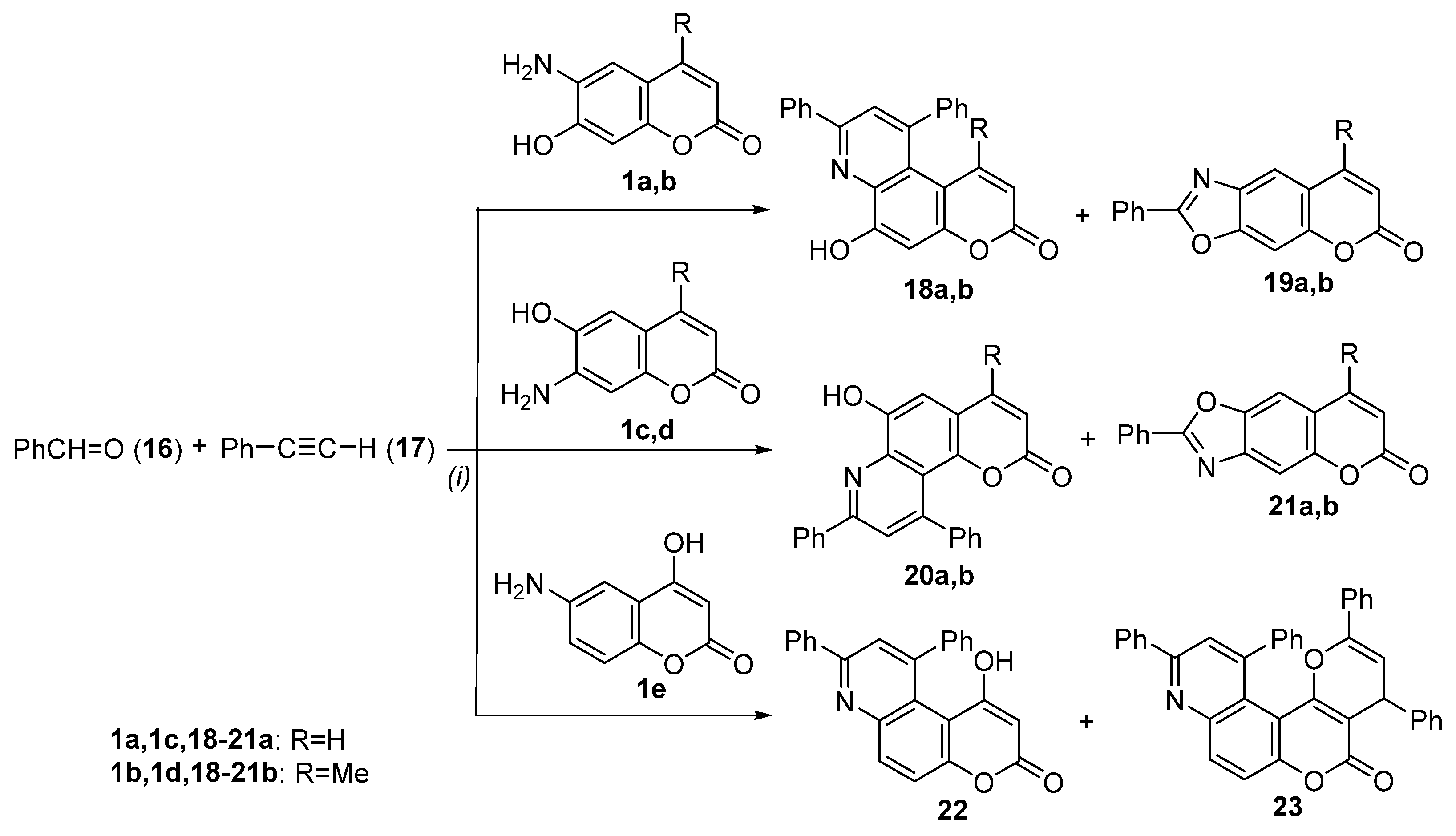
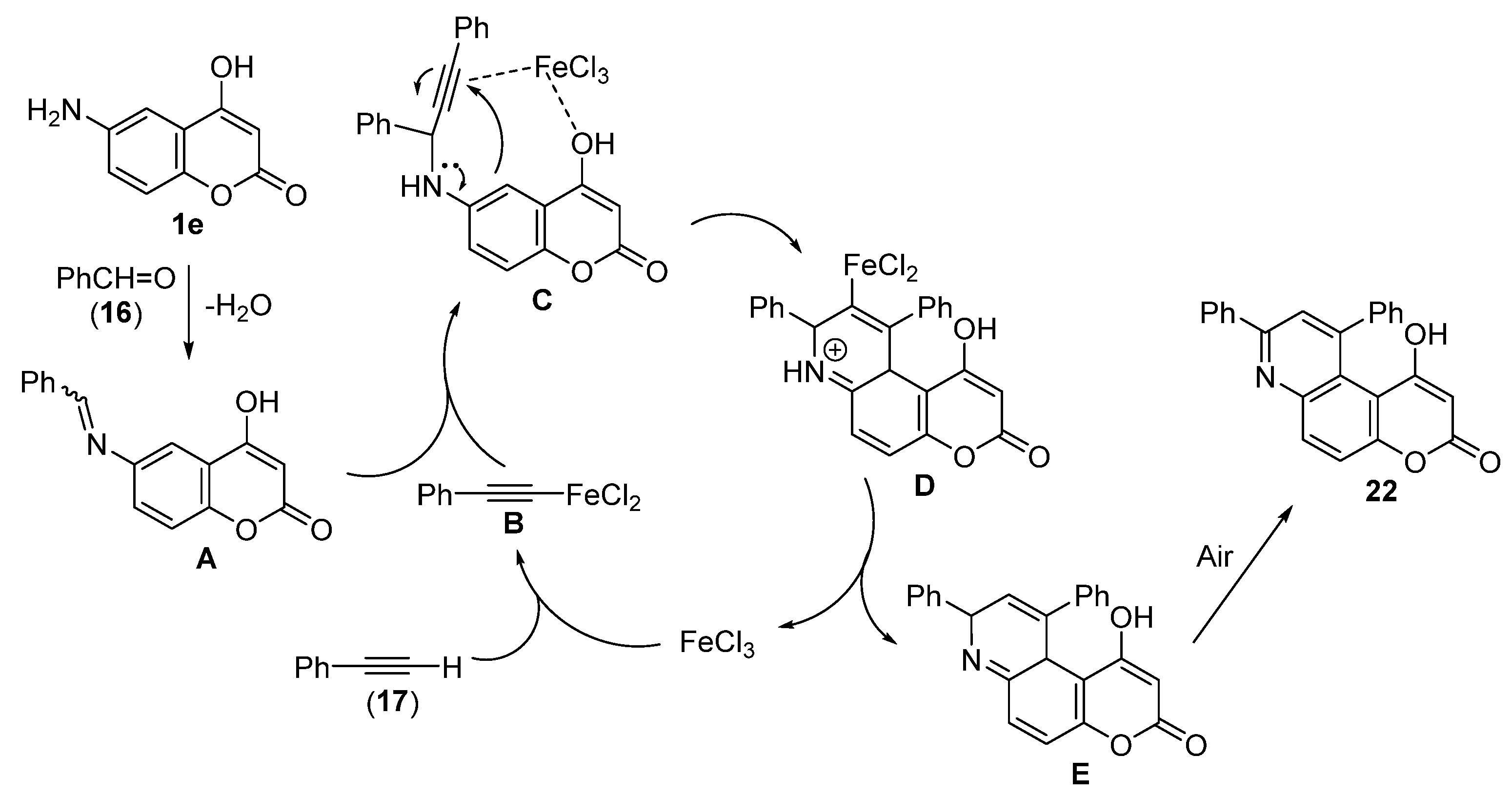
3.2.3. General Procedure for the Synthesis of Diphenyl-Substituted Pyridine Moiety of Pyranoquinolinones—Synthesis of 6-Hydroxy-8,10-diphenyl-3H-pyrano[3,2-f]quinolin-3-one (18a) and 2-Phenyl-6H-chromeno[6,7-d]oxazol-6-one (19a)
18a, Yellow Crystals, m.p. 150–152 °C (Ethyl Acetate/Hexane)
19a, Yellow Crystals, m.p. 210–211 °C (MeOH) (Lit. [66], m.p. 207–209 °C)
6-Hydroxy-1-methyl-8,10-diphenyl-3H-pyrano[3,2-f]quinolin-3-one (18b)
8-Methyl-2-phenyl-6H-chromeno[6,7-d]oxazol-6-one (19b)
6-Hydroxy-8,10-diphenyl-2H-pyrano[2,3-f]quinolin-2-one (20a)
2-Phenyl-6H-chromeno[7,6-d]oxazol-6-one (21a)
6-Hydroxy-4-methyl-8,10-diphenyl-2H-pyrano[2,3-f]quinolin-2-one (20b)
8-Methyl-2-phenyl-6H-chromeno[7,6-d]oxazol-6-one (21b)
1-Hydroxy-8,10-diphenyl-3H-pyrano[3,2-f]quinolin-3-one (22)
2,4,10,12-Tetraphenyl-4H,5H-pyrano[2’,3’:4,5]pyrano[3,2-f]quinolin-5-one (23)
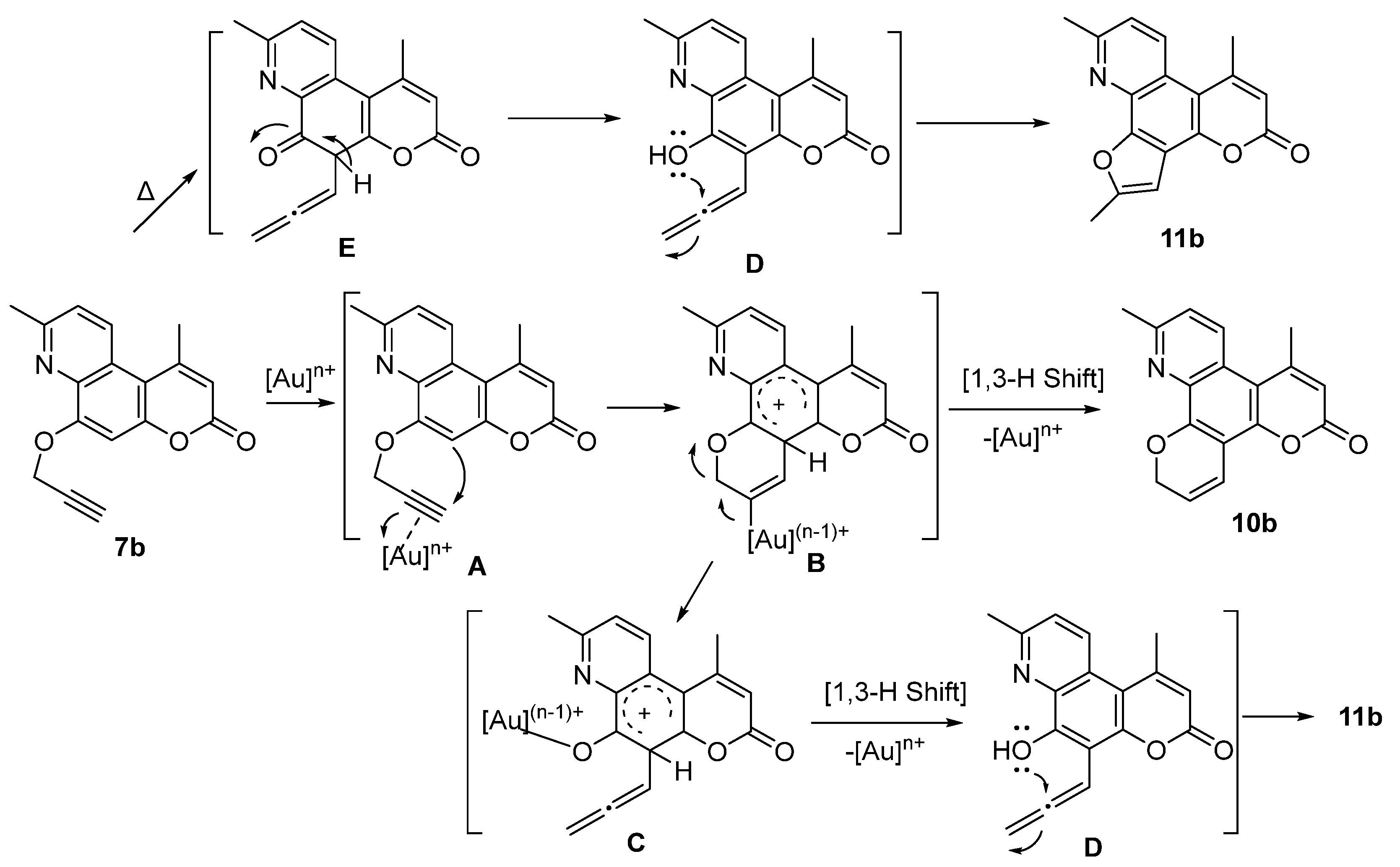
3.2.4. General Procedure for the Preparation of Pyran Derivatives via the 6-Endo-Dig Cyclization of Propargyloxy Derivatives—Synthesis of 11-Methyl-2H,6H-dipyrano[3,2-f:3’,2’-h]quinolin-6-one (10a)
10a, Light Yellow Crystals, m.p. 101–103 °C (DCM/Hexane)
8,11-Dimethyl-2H,6H-dipyrano[3,2-f:3’,2’-h]quinolin-6-one (10b)
2-Methyldipyrano[2,3-f:3’,2’-h]quinolin-6(11H)-one (12a)
2,8-Dimethyldipyrano[2,3-f:3’,2’-h]quinolin-6(11H)-one (12b)
12-Methyl-4H,5H-pyrano[2’,3’:4,5]pyrano[3,2-f]quinolin-5-one (14)
9,11-Diphenyl-2H,6H-dipyrano[3,2-f:3’,2’-h]quinolin-6-one (27a)
8-Methyl-9,11-diphenyl-2H,6H-dipyrano[3,2-f:3’,2’-h]quinolin-6-one (27b)
2,4-Diphenyldipyrano[2,3-f:3’,2’-h]quinolin-6(11H)-one (28a)
8-Methyl-2,4-diphenyldipyrano[2,3-f:3’,2’-h]quinolin-6(11H)-one (28b)
10,12-Diphenyl-2H,5H-pyrano[2’,3’:4,5]pyrano[3,2-f]quinolin-5-one (29)
3.2.5. General Procedure for the Preparation of Furan Derivatives via the 5-Exo-Dig Cyclization of Propargyloxy Derivatives—Synthesis of 2,10-Dimethyl-5H-furo[3,2-h]pyrano[3,2-f]quinolin-5-one (11a)
11a, Light Yellow Crystals, m.p. 97–99 °C (DCM/Hexane)
2,7,10-Trimethyl-5H-furo[3,2-h]pyrano[3,2-f]quinolin-5-one (11b)
2,10-Dimethyl-6H-furo[3,2-h]pyrano[2,3-f]quinolin-6-one (13a)
2,8,10-Trimethyl-6H-furo[3,2-h]pyrano[2,3-f]quinolin-6-one (13b)
2,11-Dimethyl-4H-furo[2’,3’:4,5]pyrano[3,2-f]quinolin-4-one (15)
3.3. Biological Experiments: In Vitro Assays
3.3.1. Inhibition of Linoleic Acid Peroxidation
3.3.2. Soybean Lipoxygenase Inhibition Study
3.4. Docking Studies on Soybean Lipoxygenase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Kennedy, R.; Thornes, R.D. Coumarins, Biology, Applications and Mode of Action; Wiley: Chichester, UK, 1997; p. 1. [Google Scholar]
- Fylaktakidou, K.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolaides, D. Natural and synthetic coumarin derivatives with antiinflammatory/antioxidant activity. Cur. Pharm. Des. 2004, 30, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Kontogiorgis, C.; Detsi, A.; Hadjipavlou-Litina, D. Coumarin-based drugs: A patentreview (2008–present). Expert Opin. Ther. Pat. 2012, 22, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Vekariya, R.H.; Patel, H.D. Recent advances in the synthesis of coumarin derivatives via Knoevenagel condensation: A review. Synth. Commun. 2014, 44, 2756–2788. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A natural, privileged, and versatile scaffold for bioactive compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.I.; Syed, Q.A.; Khattak, M.N.K.; Hafez, B.; Reigosa, M.J.; El-Keblawy, A. Natural product coumarins: Biological and pharmacological perspectives. Biologia 2019, 74, 863–888. [Google Scholar] [CrossRef]
- Tolba, M.S.; Abd ul-Malik, M.A.; Kamal El-Dean, A.M.; Geies, A.A.; Radwan, S.M.; Zaki, R.M.; Sayed, M.; Mohamed, S.K.; Abdel-Raheem, S.A.A. An overview on synthesis and reactions of coumarin based compounds. Curr. Chem. Lett. 2022, 11, 29–42. [Google Scholar] [CrossRef]
- Heghes, S.C.; Vostinaru, O.; Mogosan, C.; Miere, D.; Iuga, C.A.; Filip, L. Safety Profile of Nutraceuticals Rich in Coumarins: An Update. Front. Pharmacol. 2022, 13, 803338. [Google Scholar] [CrossRef]
- Miyata, R.; Shigeta, T.; Kumazawa, S.; Egi, M. Selective Syntheses of Coumarin and Benzofuran Derivatives Using Phenols and α-Methoxy-β-ketoesters. SynOpen 2023, 7, 8–16. [Google Scholar] [CrossRef]
- Akkol, E.E.; Genç, Y.; Karpuz, B.; Sanchez, E.S.; Capasso, R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Wang, Y.T.; Yan, W.; Chen, Q.L.; Huang, W.Y.; Yang, Z.; Li, X.; Wang, X.H. Inhibition viral RNP and anti-inflammatory activity of coumarins against influenza virus. Biomed. Pharm. 2017, 87, 583–588. [Google Scholar] [CrossRef]
- Al-Amiery, A.A.; Al-Majedy, Y.K.; Kadhum, A.A.; Mohamad, A.B. Novel Macromolecules Derived from Coumarin: Synthesis and Antioxidant Activity. Sci. Rep. 2015, 5, 11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.-Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.-W.; Lv, L.Z.-S.; Feng, L.S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ghobadi, E.; Ghanbarimasir, Z.; Emami, S. A review on the structures and biological activities of anti-Helicobacter pylori agents. Eur. J. Med. Chem. 2021, 223, 113669. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A Review on Coumarins as Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Liu, J.; Zhang, Y.; Hou, M.Q.; Zhang, M.Z.; Zhou, F.; Zhang, W.H. Microwave-assisted synthesis, and antifungal activity of novel coumarin derivatives: Pyrano [3,2-c] chromene-2,5-diones. Eur. J. Med. Chem. 2016, 116, 76–83. [Google Scholar] [CrossRef]
- Venkataraman, S.; Meera, R.; Ramachandran, V.; Devi, P.; Aruna, A.; Parameswari, S.P.T.; Nagarajan, K. Antioxidant and anticoagulant activity of novel n-substituted-4- methyl-5,7-dihydroxyl coumarin and its ester derivatives. Inter. J. Pharm. Rev. Res. 2014, 4, 25–32. [Google Scholar]
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic Effects of Coumarins with Different Substitution Patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef]
- Rawat, A.; Reddy, A.V.B. Recent advances on anticancer activity of coumarin derivatives. Eur. J. Med. Chem. Reports 2022, 5, 100038. [Google Scholar] [CrossRef]
- Markey, M.D.; Fu, Y.; Kelly, T.R. Synthesis of Santiagonamine. Org. Lett. 2007, 9, 3255–3257. [Google Scholar] [CrossRef]
- Patra, P.; Patra, S. Mini Review on Pyrido[2,3-c]coumarins Backbone of Santiagonamine Antibiotics. Heterocycles 2023, 106, 241–269. [Google Scholar] [CrossRef]
- Wang, X.-L.; Dou, M.; Luo, Q.; Cheng, L.-Z.; Yan, Y.-M.; Li, R.-T.; Cheng, Y.-X. Racemic alkaloids from the fungus Ganoderma cochlear. Fitoterapia 2017, 116, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Yoon, J.A.; Han, Y.T. Total Synthesis of the Natural Pyridocoumarins Goniothaline A and B. Synthesis 2019, 51, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Levrier, C.; Balastrier, M.; Beattle, K.D.; Carroll, A.R.; Martin, F.; Choomuenwai, V.; Davis, R.A. Pyridocoumarin, aristolactam and aporphine alkaloids from the Australian rainforest plant Goniothalamus Australis. Phytochem. 2013, 86, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.M.; Zhang, Q.J.; Chen, R.Y.; Yu, D.Q. Four new alkaloids from Polyalthia nemoralis (Annonaceae). J. Asian Nat. Prod. Res. 2008, 10, 656–664. [Google Scholar] [CrossRef]
- Pang, M.; Lee, J.; Jeon, J.-H.; Song, I.-S.; Han, Y.T.; Choi, M.-K. Development of a Sensitive Analytical Method of Polynemoraline C Using LCMS/MS and Its Application to a Pharmacokinetic Study in Mice. Mass Spectrom. Lett. 2021, 12, 200–205. [Google Scholar] [CrossRef]
- Gunatilaka, A.A.L.; Kingston, D.G.I.; Wijeratne, E.M.K.; Bandara, B.M.R.; Hofmann, G.A.; Johnson, R.K. Biological activity of some coumarins from Sri Lankan Rutaceae. J. Nat. Prod. 1994, 57, 518–520. [Google Scholar] [CrossRef]
- Magiatis, P.; Melliou, E.; Skaltsounis, A.L.; Mitaku, S.; Leonce, S.; Renard, P.; Pierre, A.; Atassi, G. Synthesis and cytotoxic activity of pyranocoumarins of the seselin and xanthyletin series. J. Nat. Prod. 1994, 57, 518–520. [Google Scholar] [CrossRef]
- Nivetha, R.; Bhuvaragavan, S.; Kumar, T.M.; Ramanathan, K.; Janarthanan, S. Inhibition of multiple SARS-CoV-2 proteins by an antiviral biomolecule, seselin from Aegle marmelos deciphered using molecular docking analysis. J. Biomolec. Struct. Dynam. 2021, 40, 11070–11081. [Google Scholar] [CrossRef]
- Wittayapipath, K.; Laolit, S.; Yenjai, C.; Chio-Srichan, S.; Pakarasang, M.; Tavichakorntrakool, R.; Prariyachatigul, C. Analysis of xanthyletin and secondary metabolites from Pseudomonas stutzeri ST1302 and Klebsiella pneumoniae ST2501 against Pythium insidiosum. BMC Microbiol. 2019, 19, 78. [Google Scholar] [CrossRef]
- Kang, S.Y.; Lee, K.Y.; Sung, S.H.; Park, M.J.; Kim, Y.C. Coumarins isolated from Angelica gigas inhibit acetylcholinesterase: Structure-activity relationships. J. Nat. Prod. 2001, 64, 683–685. [Google Scholar] [CrossRef]
- de Moura, N.F.; Simionatto, E.; Porto, C.; Hoelzel, S.C.S.; Dessoy, E.C.S.; Zanatta, N.; Morel, A.F. Quinoline Alkaloids, Coumarins and Volatile Constituents of Helietta longifoliata. Planta Med. 2002, 68, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.F.; Larghi, E.L. First total synthesis of (-)-(R)-geibalansin and (+)-(S)-geibalansin. Tetrahedron Asymmetry 2004, 15, 9–10. [Google Scholar] [CrossRef]
- Sapkota, B.; Devkota, H.P.; Poudel, P. Citrus maxima (Brum.) Merr. (Rutaceae): Bioactive chemical substituents and pharmacological activities. Hidawi Evid. -Based Complement. Altern. Med. 2022, 2022, 8741669. [Google Scholar] [CrossRef] [PubMed]
- Douka, M.D.; Litinas, K.E. An overview on the synthesis of pyridocoumarins with biological interest. Molecules 2022, 27, 7256. [Google Scholar] [CrossRef]
- Patra, P.; Patra, S. 4-Aminocoumarin Derivatives as Multifaceted Building Blocks for the Development of Various Bioactive Fused Coumarin Heterocycles: A Brief Review. Cur. Org. Chem. 2022, 26, 1585–1614. [Google Scholar] [CrossRef]
- Vlachou, E.-E.N.; Litinas, K.E. An Overview on Pyranocoumarins: Synthesis and Biological Activities. Curr. Org. Chem. 2019, 23, 2679–2721. [Google Scholar] [CrossRef]
- Hsieh, W.-C.; Lin, C.-H.; Yang, Y.-J.; Yang, D.-Y. Multicomponent synthesis of pyrano[2,3-c] coumarins. RSC Adv. 2018, 8, 39162–39169. [Google Scholar] [CrossRef]
- Vlachou, E.-E.N.; Fotopoulos, I.; Gabriel, C.; Pontiki, E.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Synthesis and biological evaluation of fused dipyranoquinolinones as inhibitors of acetylcholinesterase with antioxidant properties. Eur. J. Med. Chem. Reports 2022, 5, 100063. [Google Scholar] [CrossRef]
- Symeonidis, T.S.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Synthesis Through Three-Component Reactions Catalyzed by FeCl3 of Fused Pyridocoumarins as Inhibitors of Lipid Peroxidation. J. Heterocycl. Chem. 2014, 51, 642–647. [Google Scholar] [CrossRef]
- Symeonidis, T.S.; Litinas, K.E. Synthesis of methyl substituted [5,6]- and [7,8]-fused pyridocoumarins via the iodine-catalyzed reaction of aminocoumarins with n-butyl vinyl ether. Tetrahedron Lett. 2013, 54, 6517–6519. [Google Scholar] [CrossRef]
- Symeonidis, T.S.; Lykakis, I.N.; Litinas, K.E. Synthesis of quinolines and fused pyridocoumarins from N-propargylanilines or propargylaminocoumarins by catalysis with gold nanoparticles supported on TiO2. Tetrahedron 2013, 69, 4612–4616. [Google Scholar] [CrossRef]
- Symeonidis, T.S.; Kallitsakis, M.G.; Litinas, K.E. Synthesis of [5,6]-fused pyridocoumarins through aza-Claisen rearrangement of 6-propargylaminocoumarins. Tetrahedron Lett. 2011, 52, 5452–5455. [Google Scholar] [CrossRef]
- Gautam, D.R.; Protopappas, J.; Fylaktakidou, K.C.; Litinas, K.E.; Nicolaides, D.N.; Tsoleridis, C.A. Unexpected one-pot synthesis of new polycyclic coumarin[4,3-c]pyridine derivatives via a tandem hetero-Diels-Alder and 1,3-dipolar cycloaddition reaction. Tetrahedron Lett. 2009, 50, 448–451. [Google Scholar] [CrossRef]
- Galariniotou, E.; Fragos, V.; Makri, A.; Litinas, K.E.; Nicolaides, D.N. Synthesis of novel pyridocoumarins and benzo- fused 6-azacoumarins. Tetrahedron 2007, 63, 8298–8304. [Google Scholar] [CrossRef]
- Vlachou, E.-E.N.; Balalas, T.D.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Douka, M.D. 2,9-Dimethyl-4H-oxazolo[5′,4′:4,5]pyrano[3,2-f]quinoline-4-one. Molbank 2023, 2023, M1591. [Google Scholar] [CrossRef]
- Vlachou, E.-E.N.; Gabriel, C.; Litinas, K.E. One-pot Synthesis of Fused Dipyranocoumarins from Dihydroxycoumarins and Propargyl Chlorides under Microwave Irradiation. J. Heterocyclic Chem. 2019, 56, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Litinas, K.E.; Symeonidis, T.S. Convenient synthesis of fused pyrano[3,2-h]- and furo[3,2-h]benzo[f]coumarins from naphthalene-2,3-diols. Tetrahedron 2010, 66, 1289–1293. [Google Scholar] [CrossRef]
- Tsoukka, M.; Litinas, K.E.; Nicolaides, D.N.; Hadjipavlou-Litina, D.J. Synthesis and biological evaluation of new benzo[f]furo[2,3-h]- and benzo[f]pyrano[2,3-h]coumarin derivatives. J. Heterocyclic Chem. 2007, 44, 529–534. [Google Scholar] [CrossRef]
- Baldoumi, V.; Gautam, D.R.; Litinas, K.E.; Nicolaides, D.N. Convenient synthesis of linear pyrano[3,2-g]-. [2,3-g]- and angular pyrano[3,2-f]coumarins from 4-[(1,1-dimethyl-2-propynyl)oxy]phenol. Tetrahedron 2006, 62, 8016–8020. [Google Scholar] [CrossRef]
- Nicolaides, D.N.; Gautam, D.R.; Litinas, K.E.; Hadjipavlou-Litina, D.J.; Fylaktakidou, K.C. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of some benzo[l]khellactone derivatives and analogues. Eur. J.Med. Chem. 2004, 39, 323–332. [Google Scholar] [CrossRef]
- Nicolaides, D.N.; Gautam, D.R.; Litinas, K.E.; Papamehael, T. Synthesis of some 3,4-dihydro-2H-benzo[f]pyrano[2,3-h]chromen-6-one derivatives. J. Chem. Soc. Perkin Trans 1 2002, 33, 1455–1460. [Google Scholar] [CrossRef]
- Lin, S.T.; Yang, F.-M.; Yang, H.-J.; Huang, K.-F. Preparation of amino- and formylaminocoumarins by selective hydrogenation of nitrocoumarins. J. Chem. Res. 1995, 27, 372–373. [Google Scholar] [CrossRef]
- Soares, A.M.S.; Costa, S.P.G.; Sameiro, M.; Gonçalves, T. Oxazole light triggered protecting groups: Synthesis and photolysis of fused heteroaromatic conjugates. Tetrahedron 2010, 66, 8189–8195. [Google Scholar] [CrossRef]
- De, P. Efficient reduction of nitroarenes with SnCl2 in ionic liquid. Synlett 2004, 10, 1835–1837. [Google Scholar] [CrossRef]
- Uliassi, E.; Fiorani, G.; Krauth-Siegel, R.L.; Bergamini, C.; Fato, R.; Bianchini, G.; Menéndez, J.C.; Molina, M.T.; López-Montero, E.; Falchi, F.; et al. Crassiflorone derivatives that inhibit Trypanosoma brucei glyceraldehyde-3-phosphate dehydrogenase (TbGAPDH) and Trypanosoma cruzi trypanothione reductase (TcTR) and display trypanocidal activity. Eur. J. Med. Chem. 2017, 141, 138–148. [Google Scholar] [CrossRef]
- Neetha, M.; Aneeja, T.; Afsina, C.M.A.; Anilkumar, G. An Overview of Ag-catalyzed Synthesis of Six-membered Heterocycles. ChemCatChem 2020, 12, 5330–5358. [Google Scholar] [CrossRef]
- Efe, C.; Lykakis, I.N.; Stratakis, M. Gold nanoparticles supported on TiO2 catalyse the cycloisomerisation/oxidative dimerisation of aryl propargyl ethers. Chem. Commun. 2011, 47, 803–805. [Google Scholar] [CrossRef]
- Praveen, C.; Dupeux, A.; Michelet, V. Catalytic Gold Chemistry: From Simple Salts to Complexes for Regioselective C–H Bond Functionalization. Chem. Eur. J. 2021, 27, 10495–10532. [Google Scholar] [CrossRef] [PubMed]
- Arcadi, A.; Ciogli, A.; Fabrizi, G.; Fochetti, A.; Franzini, R.; Ghirga, F.; Goggiamani, A.; Iazzetti, A. Synthesis of pyrano[2,3-f]chromen-2-ones vs. pyrano[3,2-g]chromen-2-ones through site controlled gold-catalyzed annulations. Org. Biomol. Chem. 2019, 17, 10065–10072. [Google Scholar] [CrossRef]
- Lau, V.M.; Pfalzgraff, W.C.; Markland, T.E.; Kanan, M.W. Electrostatic Control of Regioselectivity in Au(I)- Catalyzed Hydroarylation. J. Am. Chem. Soc. 2017, 139, 4035–4041. [Google Scholar] [CrossRef]
- Menon, R.S.; Findlay, A.D.; Bissember, A.C.; Banwell, M.G. The Au(I)-Catalyzed Intramolecular Hydroarylation of Terminal Alkynes Under Mild Conditions: Application to the Synthesis of 2H-Chromenes, Coumarins, Benzofurans, and Dihydroquinolines. J. Org. Chem. 2009, 74, 8901–8903. [Google Scholar] [CrossRef]
- Ishii, H.; Ishikawa, T.; Takeda, S.; Ueki, S.; Suzuki, M. Cesium fluoride mediated Claisen rearrangement of aryl propargyl ether. Exclusive formation of 2-methylarylfuran and its availability as a masked salicylaldehyde. Chem. Pharm. Bull. 1992, 40, 1148–1153. [Google Scholar] [CrossRef] [Green Version]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furstner, A.; Davies, P.W. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, E.-E.N.; Armatas, G.S.; Litinas, K.E. Synthesis of Fused Oxazolocoumarins from o-Hydroxynitrocoumarins and Benzyl Alcohol Under Gold Nanoparticles or FeCl3. J. Heterocyclic Chem. 2017, 53, 2447–2453. [Google Scholar] [CrossRef]
- Ren, Q.; Kang, J.; Li, M.; Yuan, L.; Chen, R.; Wang, L. Regioselective Access to Structurally Diverse Coumarin Analogues through Iron-Catalysed Annulation Reactions. Eur. J. Org. Chem. 2017, 2017, 5566–5571. [Google Scholar] [CrossRef]
- Kasthuri, J.K.; Singh, J.S.; Thripuram, V.D.; Gundabolu, U.R.; Ala, V.; Kolla, J.N.; Jayaprakash, V.; Ahsan, M.J.; Bollikolla, H.B. Synthesis, Characterization, Docking and Study of Inhibitory Action of Some Novel C-Alkylated Chalcones on 5-LOX Enzyme. ChemistrySelect 2017, 2, 8771–8778. [Google Scholar] [CrossRef]
- Kostopoulou, I.; Tzani, A.; Polyzos, N.-I.; Karadendrou, M.A.; Kritsi, E.; Pontiki, E.; Liargkova, T.; Hadjipavlou-Litina, D.; Zoumpoulakis, P.; Detsi, A. Exploring the 2′-Hydroxy-Chalcone Framework for the Development of Dual Antioxidant and Soybean Lipoxygenase Inhibitory Agents. Molecules 2021, 26, 2777. [Google Scholar] [CrossRef]
- Mavridis, E.; Bermperoglou, E.; Pontiki, E.; Hadjipavlou-Litina, D. 5-(4H)-Oxazolones and Their Benzamides as Potential Bioactive Small Molecules. Molecules 2020, 25, 3173. [Google Scholar] [CrossRef]
- Mantzanidou, M.; Pontiki, E.; Hadjipavlou-Litina, D. Pyrazoles and Pyrazolines as Anti-Inflammatory Agents. Molecules 2021, 26, 3439. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Šali, A.B.T.-M. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. Available online: http://www.jcheminf.com/content/3/1/33 (accessed on 7 October 2011). [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes. 2012, 5, 367. Available online: http://www.biomedcentral.com/1756-0500/5/367 (accessed on 23 July 2012). [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reacting Compounds | Reaction Conditions | Product (Yield, %) |
|---|---|---|---|
| 1 | 1a, 2 (3 equiv.) | A: I2 (10 mol%), CH3CN, reflux, 1 h | 3a (94) |
| 2 | 1b, 2 (3 equiv.) | A | 3b (86) |
| 3 | 1c, 2 (3 equiv.) | A | 4a (98) |
| 4 | 1d, 2 (3 equiv.) | A | 4b (87) |
| 5 | 1e, 2 (3 equiv.) | A | 5 (90) |
| 6 | 3a, 6 (1.1 equiv.) | B: Cs2CO3 (1.1 equiv.), acetone, MW, 100 °C, 10 min | 7a (99) |
| 7 | 3b, 6 (1.1 equiv.) | B | 7b (93) |
| 8 | 4a, 6 (1.1 equiv.) | B | 8a (97) |
| 9 | 4b, 6 (1.1 equiv.) | B | 8b (93) |
| 10 | 5, 6 (1.1 equiv.) | B | 9 (99) |
| 11 | 7a | C: Au/TiO2 (4 mol%), PhCl, MW, 180 °C, 2 h | 10a (96) |
| 12 | 7b | C | 10b (93) |
| 13 | 8a | C | 12a (96) |
| 14 | 8b | C | 12b (91) |
| 15 | 9 | C | 14 (94) |
| 16 | 7a | D: PhCl, MW, 180 °C, 2.5 h | 11a (90) |
| 17 | 7b | D | 11b (98) |
| 18 | 8a | D | 13a (95) |
| 19 | 8b | D | 13b (93) |
| 20 | 9 | D | 15 (93) |
| Entry | Conditions | Products (Yield %) |
|---|---|---|
| 1 | Au/TiO2 (4 mol%), DCE, MW, 140 °C, 3 h | - |
| 2 | BF3·Et2O, DMF, MW, 200 °C, 1 h | - |
| 3 | AgNO3 (10 mol%), DCE, MW, 140 °C, 1 h | - |
| 4 | Au/TiO2 (4 mol%), PhCl, MW, 180 °C, 2 h | 10b (93) |
| 5 | TiO2 (4 mol%), PhCl, MW, 180 °C, 3 h | 11b (99) |
| 6 | PhCl, MW, 180 °C, 2.5 h | 11b (98) |
| 7 | PhCl, 120 °C, 24 h | 11b (10), 7b (90) |
| 8 | AuCl3 (4 mol%), PhCl, MW, 180 °C, 30 min | 10b (90) |
| 9 | AuCl (4 mol%), PhCl, MW, 180 °C, 1 h | 10b (50), 11b (50) |
| Entry | Reacting Compounds | Reaction Conditions | Products (Yield, %) |
|---|---|---|---|
| 1 | 1a, 16 (1.1 equiv.), 17 (1.1 equiv.) | E: FeCl3·6H2O (10 mol%), toluene, reflux, 24 h | 18a (66), 19a (32) |
| 2 | 1b, 16 (1.1 equiv.), 17 (1.1 equiv.) | E | 18b (62), 19b (35) |
| 3 | 1c, 16 (1.1 equiv.), 17 (1.1 equiv.) | E | 20a (65), 21a (29) |
| 4 | 1d, 16 (1.1 equiv.), 17 (1.1 equiv.) | E | 20b (63), 21b (33) |
| 5 | 1e, 16 (1.1 equiv.), 17 (1.1 equiv.) | E | 22 (82), 23 (10) |
| 6 | 18a, 6 (1.1 equiv.) | F: Cs2CO3 (1.1 equiv.), acetone, MW, 100 °C, 5 min | 24a (99) |
| 7 | 18b, 6 (1.1 equiv.) | F | 24b (92) |
| 8 | 20a, 6 (1.1 equiv.) | F | 25a (98) |
| 9 | 20b, 6 (1.1 equiv.) | F | 25b (95) |
| 10 | 22, 6 (1.1 equiv.) | F | 26 (98) |
| 11 | 24a | C: Au/TiO2 (4 mol%), PhCl, MW, 180 °C, 2 h | 27a (97) |
| 12 | 24b | C | 27b (99) |
| 13 | 25a | C | 28a (96) |
| 14 | 25b | C | 28b (93) |
| 15 | 26 | C | 29 (92) |
| Entry | Compound | LOX at 100 μM (%) or IC50 (μM) | LP at 100 μM (%) |
|---|---|---|---|
| 1 | 10a | no | no |
| 2 | 10b | no | 66% |
| 3 | 12a | 40% | no |
| 4 | 12b | 100 μM | no |
| 5 | 14 | 100 μM | 19% |
| 6 | 27a | 82.5 μM | 32% |
| 7 | 27b | 60 μM | no |
| 8 | 28a | 10 μM | 41% |
| 9 | 28b | 100 μM | 70% |
| 10 | 29 | 85 μM | 29% |
| 11 | NDGA | 0.45 μM | |
| 12 | Trolox | 91% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlachou, E.-E.N.; Pontiki, E.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Synthesis and Biological Evaluation of Substituted Fused Dipyranoquinolinones. Organics 2023, 4, 364-385. https://doi.org/10.3390/org4030027
Vlachou E-EN, Pontiki E, Hadjipavlou-Litina DJ, Litinas KE. Synthesis and Biological Evaluation of Substituted Fused Dipyranoquinolinones. Organics. 2023; 4(3):364-385. https://doi.org/10.3390/org4030027
Chicago/Turabian StyleVlachou, Evangelia-Eirini N., Eleni Pontiki, Dimitra J. Hadjipavlou-Litina, and Konstantinos E. Litinas. 2023. "Synthesis and Biological Evaluation of Substituted Fused Dipyranoquinolinones" Organics 4, no. 3: 364-385. https://doi.org/10.3390/org4030027