
Facile Photochemical/Thermal Assisted Hydration of Alkynes Catalysed under Aqueous Media by a Chalcogen Stabilized, Robust, Economical, and Reusable Fe3Se2(CO)9 Cluster
 , , ,
, , ,
Abstract
:1. Introduction
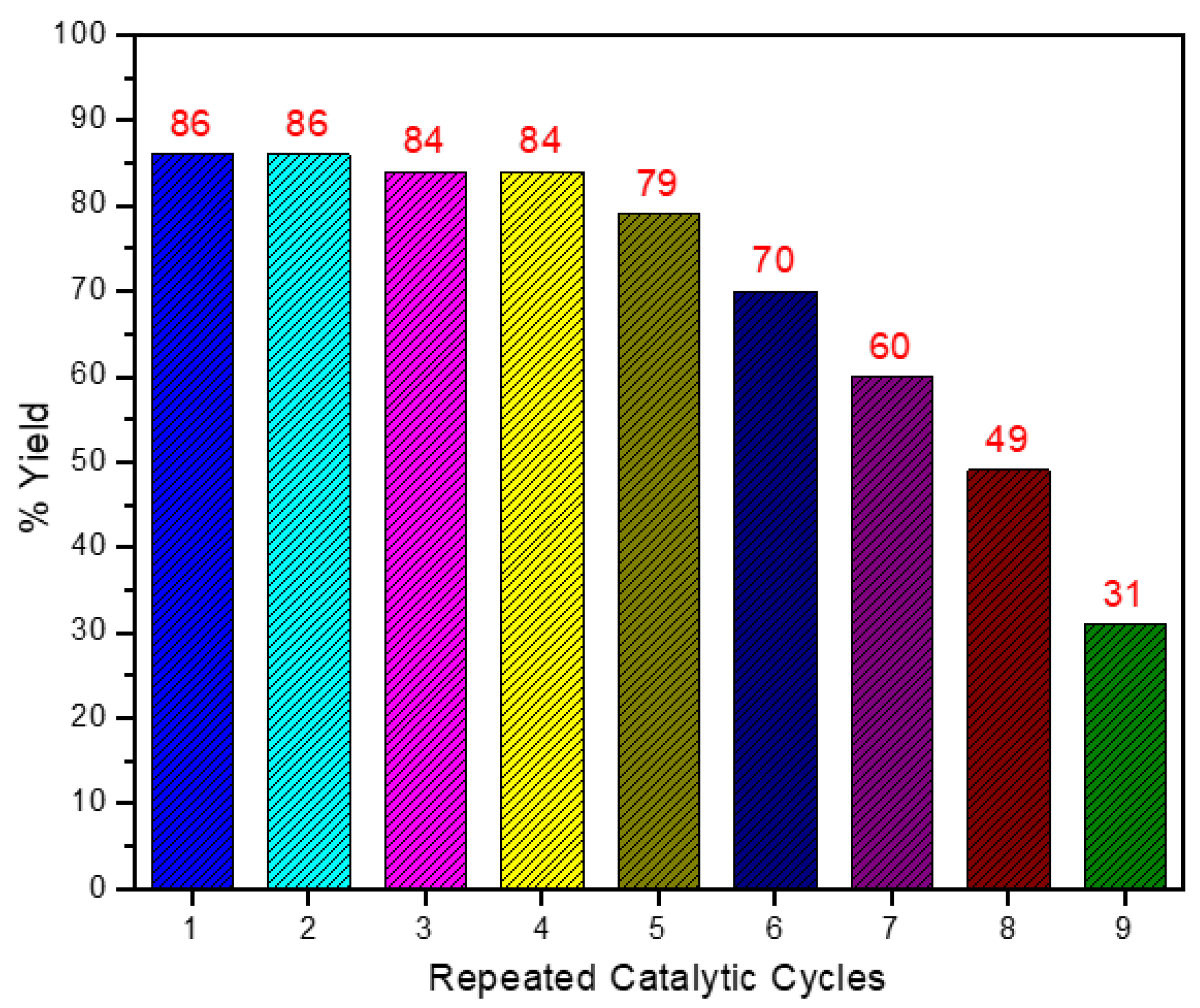
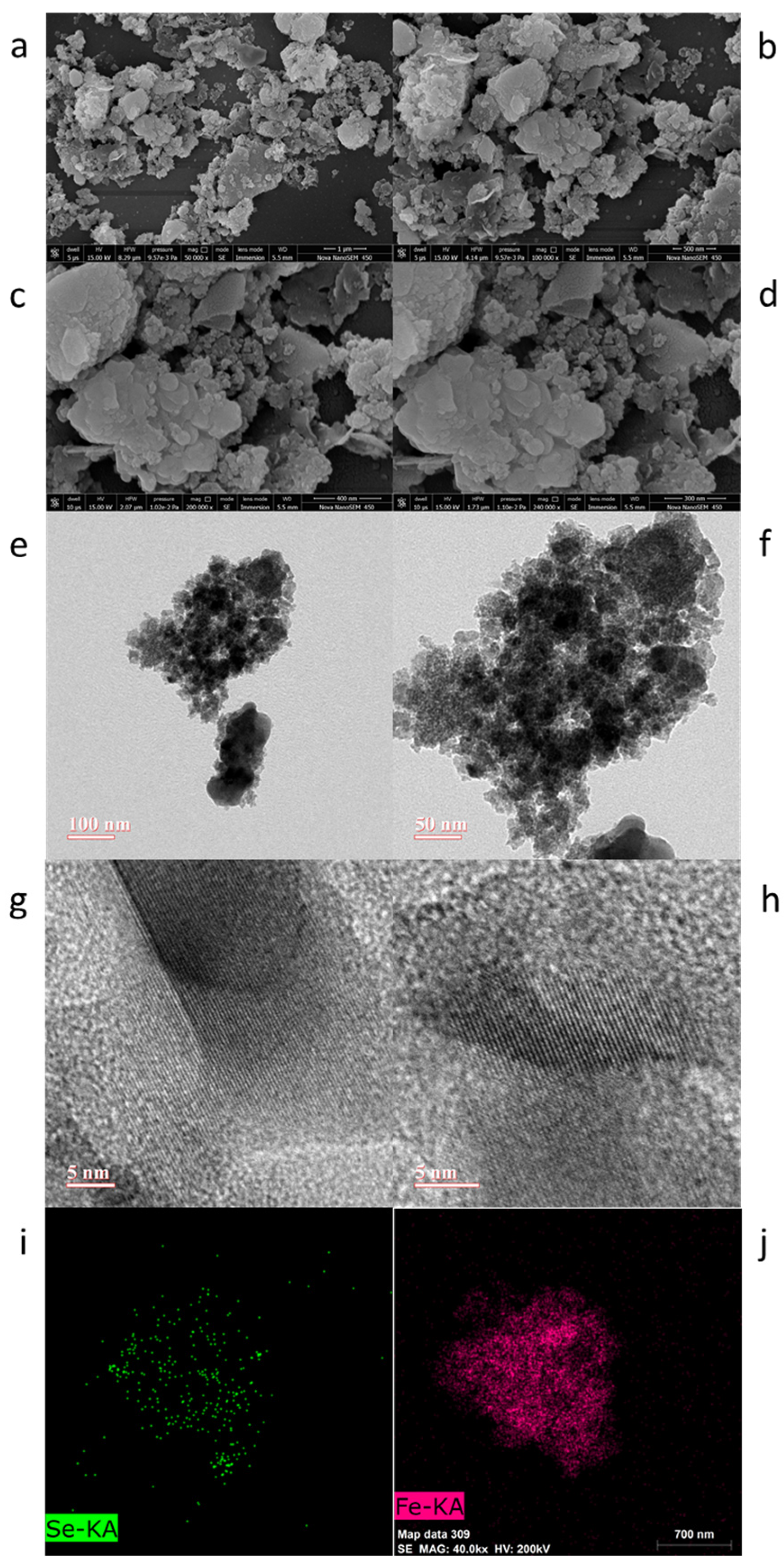
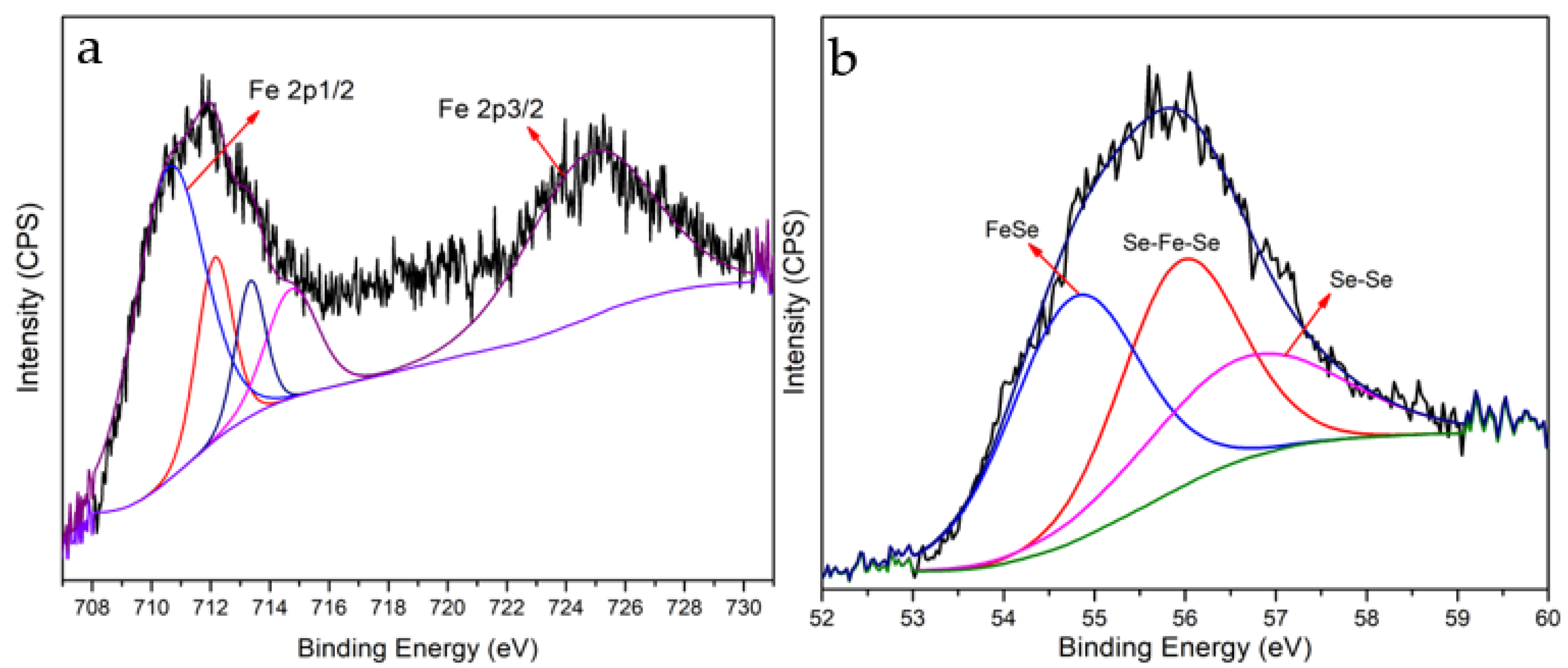
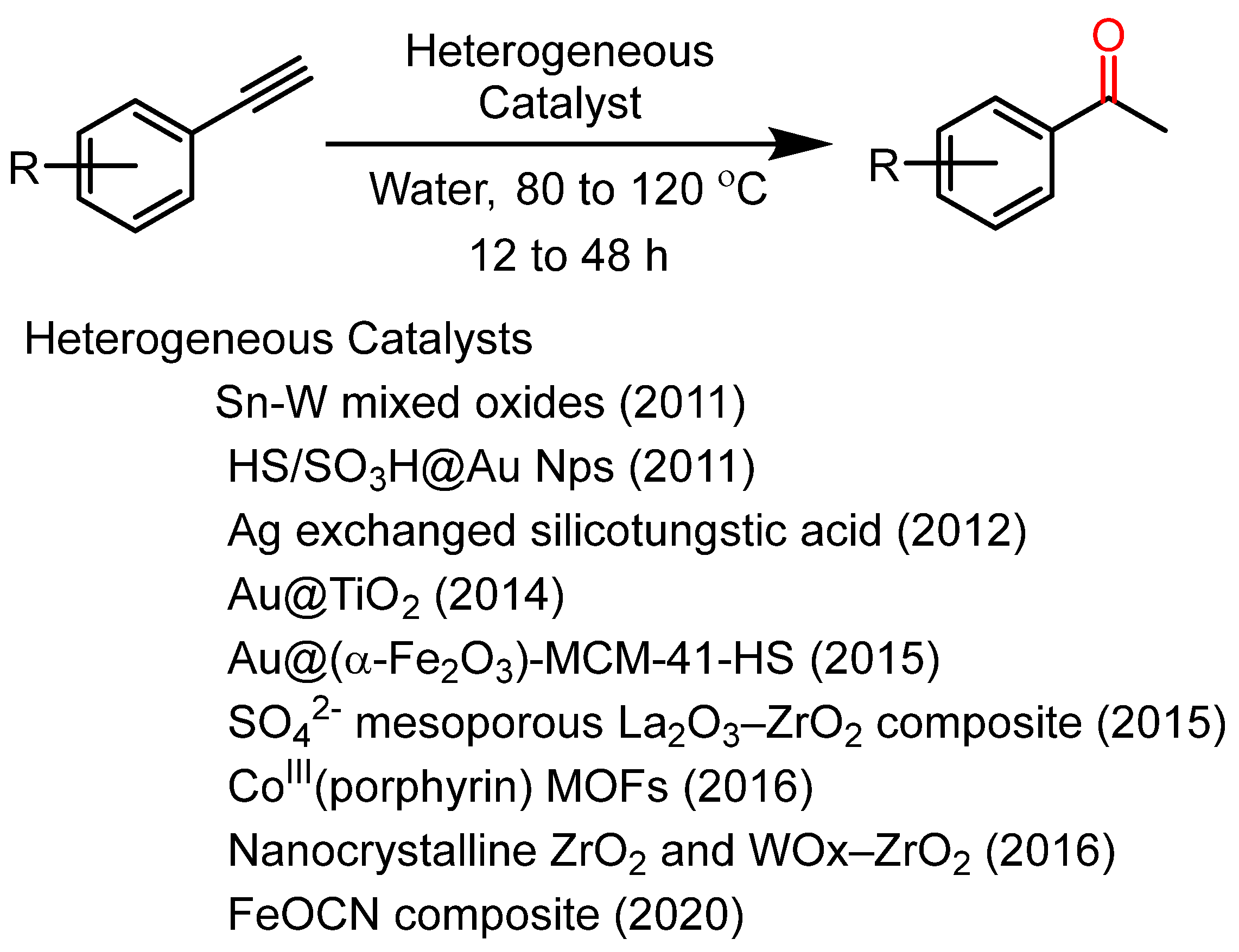
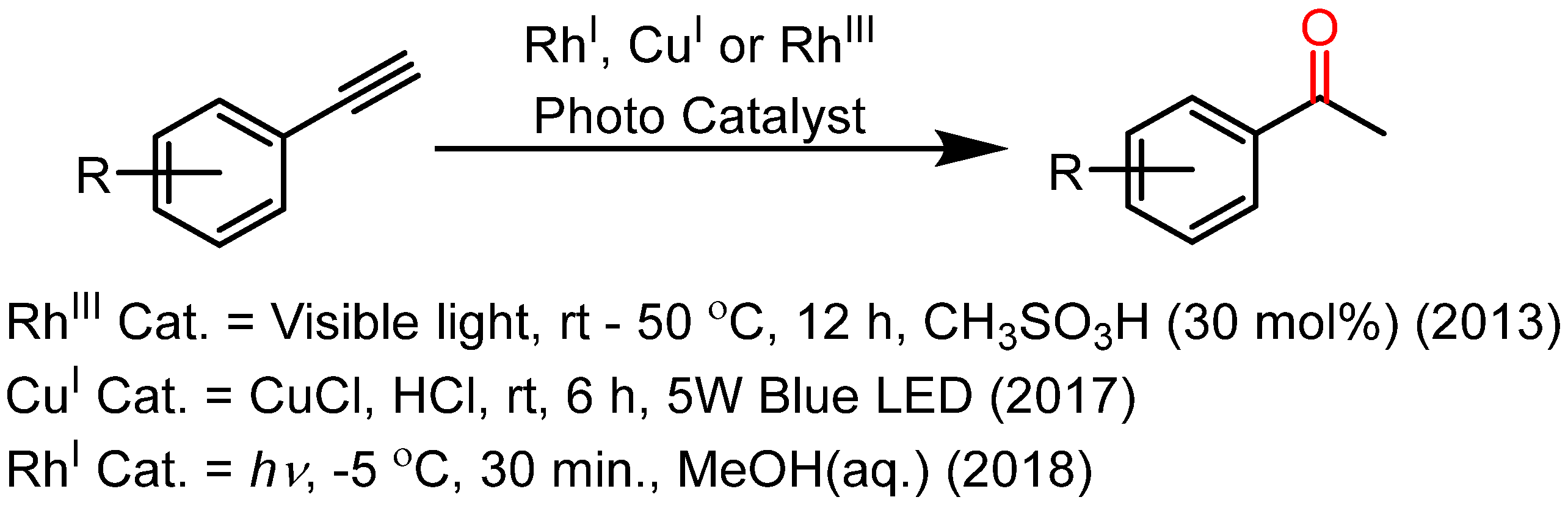
2. Results and Discussion
3. Materials and Methods
3.1. Photochemical Condition
3.2. Thermal Condition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hintermann, L.; Labonne, A. Catalytic Hydration of Alkynes and Its Application in Synthesis. Synthesis 2007, 8, 1121. [Google Scholar] [CrossRef]
- Salvio, R.; Basseti, M. Sustainable hydration of alkynes promoted by first row transition metal complexes. Background, highlights and perspectives. Inorganica Chim. Acta 2021, 522, 120288. [Google Scholar] [CrossRef]
- Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Highly Efficient AuI-Catalyzed Hydration of Alkynes. Angew. Chem. 2002, 114, 4745. [Google Scholar] [CrossRef]
- Ali, M.; Srivastava, A.K.; Siangwata, S.; Smith, G.S.; Joshi, R.K. Photo induced alkyne hydration reactions mediated by a water soluble, reusable Rhodium (I) catalyst. Catal. Commun. 2018, 115, 78. [Google Scholar] [CrossRef]
- Halpern, J.; James, B.R.; Kemp, A.L.W. Catalysis of The Hydration Of Acetylenic Compounds By Ruthenium(III) Chloride. J. Am. Chem. Soc. 1961, 83, 4097. [Google Scholar] [CrossRef]
- Halpern, J.; James, B.R.; Kemp, A.L.W. Formation and Properties of Some Chlorocarbonyl Complexes of Ruthenium(II) and Ruthenium(III). J. Am. Chem. Soc. 1966, 88, 5142. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Incarvito, C.D.; Rheingold, A.L. Combined Effects of Metal and Ligand Capable of Accepting a Proton or Hydrogen Bond Catalyze Anti-Markovnikov Hydration of Terminal Alkynes. Angew. Chem. Int. Ed. 2001, 40, 3884. [Google Scholar] [CrossRef]
- Tokunaga, M.; Wakatsuki, Y. The First Anti-Markovnikov Hydration of Terminal Alkynes: Formation of Aldehydes Catalyzed by a Ruthenium(II)/Phosphane Mixture. Angew. Chem. Int. Ed. 1998, 37, 2867. [Google Scholar] [CrossRef]
- Suzuki, T.; Tokunaga, M.; Wakatsuki, Y. Ruthenium Complex-Catalyzed anti-Markovnikov Hydration of Terminal Alkynes. Org. Lett. 2001, 3, 735. [Google Scholar] [CrossRef]
- Tokunaga, M.; Suzuki, T.; Koga, N.; Fukushima, T.; Horiuchi, A.; Wakatsuki, Y. Ruthenium-Catalyzed Hydration of 1-Alkynes to Give Aldehydes: Insight into anti-Markovnikov Regiochemistry. J. Am. Chem. Soc. 2001, 123, 11917. [Google Scholar] [CrossRef]
- Hiscox, W.; Jennings, P.W. Synthesis and reactions of nickel and palladium carbon-bound enolate complexes. Organometallics 1990, 9, 1997. [Google Scholar] [CrossRef]
- Baidossi, W.; Lahav, M.; Blum, J. Hydration of Alkynes by a PtCl4−CO Catalyst. J. Org. Chem. 1997, 62, 669. [Google Scholar] [CrossRef]
- Francisco, L.W.; Moreno, D.A.; Atwood, J.D. Synthesis, Characterization, and Reaction Chemistry of PtCl2[P(m-C6H4SO3Na)3]2, an Alkyne Hydration Catalyst. Organometallics 2001, 20, 4237. [Google Scholar] [CrossRef]
- Fukuda, Y.; Utimoto, K. Effective transformation of unactivated alkynes into ketones or acetals with a gold(III) catalyst. J. Org. Chem. 1991, 56, 3729. [Google Scholar] [CrossRef]
- Fukuda, Y.; Utimoto, K. Efficient transformation of methyl propargyl ethers into α, β-unsaturated ketones. Bull. Chem. Soc. Jpn. 1991, 64, 2013. [Google Scholar] [CrossRef]
- Imi, K.; Imai, K.; Utimoto, K. Regioselective hydration of alkynones by palladium catalysis. Tetrahedron Lett. 1987, 28, 3127. [Google Scholar] [CrossRef]
- Meier, K.; Marsella, J.A. Hydration of acetylenic compounds without using mercury. J. Mol. Catal. 1993, 78, 31. [Google Scholar] [CrossRef]
- Ghosh, N.; Nayak, S.; Sahoo, A.K. Gold-Catalyzed Regioselective Hydration of Propargyl Acetates Assisted by a Neighboring Carbonyl Group: Access to α-Acyloxy Methyl Ketones and Synthesis of (±)-Actinopolymorphol B. J. Org. Chem. 2011, 76, 500. [Google Scholar] [CrossRef]
- Leyva, A.; Corma, A. Isolable Gold(I) Complexes Having One Low-Coordinating Ligand as Catalysts for the Selective Hydration of Substituted Alkynes at Room Temperature without Acidic Promoters. J. Org. Chem. 2009, 74, 2067. [Google Scholar] [CrossRef]
- Nun, P.; Ramón, R.S.; Gaillard, S.; Nolan, S.P. Efficient silver-free gold (I)-catalyzed hydration of alkynes at low catalyst loading. J. Organomet. Chem. 2011, 696, 7. [Google Scholar] [CrossRef]
- Li, F.; Wang, N.; Lu, L.; Zhu, G. Regioselective Hydration of Terminal Alkynes Catalyzed by a Neutral Gold(I) Complex [(IPr)AuCl] and One-Pot Synthesis of Optically Active Secondary Alcohols from Terminal Alkynes by the Combination of [(IPr)AuCl] and Cp*RhCl[(R,R)-TsDPEN]. J. Org. Chem. 2015, 80, 3538. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Zotto, A.D.; Tarantelli, F.; Zuccaccia, D. Hydration and alkoxylation of alkynes catalyzed by NHC–Au–OTf. Green Chem. 2018, 20, 2125. [Google Scholar] [CrossRef]
- Thuong, M.B.T.; Mann, A.; Wagner, A. Mild chemo-selective hydration of terminal alkynes catalysed by AgSbF6. Chem. Commun. 2012, 48, 434. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.P.; Pastel, M. FeCl3 H2O: A specific system for arylacetylene hydration. J. Organomet. Chem. 1996, 522, 303. [Google Scholar] [CrossRef]
- Wu, X.; Bezier, D.; Darcel, C. Development of the First Iron Chloride Catalyzed Hydration of Terminal Alkynes. Adv. Synth. Catal. 2009, 351, 367. [Google Scholar] [CrossRef]
- Antonino, J.R.C.; Perez, A.L.; Corma, A. Regioselective hydration of alkynes by iron (III) Lewis/Brønsted catalysis. Chem. Eur. J. 2012, 18, 11107. [Google Scholar] [CrossRef]
- Bassetti, M.; Ciceri, S.; Lancia, F.; Pasquini, C. Hydration of aromatic terminal alkynes catalyzed by iron (III) sulfate hydrate under chlorine-free conditions. Tetrahedron Lett. 2014, 55, 1608. [Google Scholar] [CrossRef]
- Hou, S.; Yang, H.; Cheng, B.; Zhai, H.; Li, Y. Cobaloxime-catalyzed hydration of terminal alkynes without acidic promoters. Chem. Commun. 2017, 53, 6926. [Google Scholar] [CrossRef]
- Tachinami, T.; Nishimura, T.; Ushimaru, R.; Noyori, R.; Naka, H. Hydration of terminal alkynes catalyzed by water-soluble cobalt porphyrin complexes. J. Am. Chem. Soc. 2013, 135, 50. [Google Scholar] [CrossRef]
- Jin, X.; Oishi, T.; Yamaguchi, K.; Mizuno, N. Heterogeneously catalyzed efficient hydration of alkynes to ketones by tin–tungsten mixed oxides. Chem. Eur. J. 2011, 17, 1261. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, W.; Li, H. Water-Medium and Solvent-Free Organic Reactions over a Bifunctional Catalyst with Au Nanoparticles Covalently Bonded to HS/SO3H Functionalized Periodic Mesoporous Organosilica. J. Am. Chem. Soc. 2011, 133, 11632. [Google Scholar] [CrossRef]
- Venkateswara Rao, K.T.; Sai Prasad, P.S.; Lingaiah, N. Solvent-free hydration of alkynes over a heterogeneous silver exchanged silicotungstic acid catalyst. Green Chem. 2012, 14, 1507. [Google Scholar] [CrossRef]
- Liang, S.; Jasinski, J.; Hammond, G.B.; Xu, B. Supported gold nanoparticle-catalyzed hydration of alkynes under basic conditions. Org. Lett. 2015, 17, 162. [Google Scholar] [CrossRef]
- Rostamizadeh, H.; Estiri, S.; Azad, M. Au anchored to (α-Fe2O3)-MCM-41-HS as a novel magnetic nanocatalyst for water-medium and solvent-free alkyne hydration. Catal. Commun. 2014, 57, 29. [Google Scholar] [CrossRef]
- Zhao, Z.; Ran, J. Sulphated mesoporous La2O3–ZrO2 composite oxide as an efficient and reusable solid acid catalyst for alkenylation of aromatics with phenylacetylene. Appl. Catal. A Gen. 2015, 503, 77. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Z.; Chen, Y.; Lin, W. Highly Efficient Cooperative Catalysis by CoIII(Porphyrin) Pairs in Interpenetrating Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2016, 55, 13739. [Google Scholar] [CrossRef]
- Gonell, F.; Portehault, D.; Julián-López, K.; Vallé, B.; Sanchez, C.; Corma, A. One step microwave-assisted synthesis of nanocrystalline WO x–ZrO 2 acid catalysts. Catal. Sci. Technol. 2016, 6, 8257. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Ali, M.; Siangwata, S.; Satrawala, N.; Smith, G.S.; Joshi, R.K. Multitasking FeOCN Composite as an Economic, Heterogeneous Catalyst for 1-Octene Hydroformylation and Hydration Reactions. Asian J. Org. Chem. 2020, 9, 377. [Google Scholar] [CrossRef]
- Vasudevan, A.; Verzal, M.K. Neutral, metal-free hydration of alkynes using microwave irradiation in superheated water. SYNLETT 2004, 4, 631. [Google Scholar] [CrossRef]
- Ali, M.; Srivastava, A.K.; Joshi, R.K. Metal/catalyst/reagent free hydration of alkynes up to gram scale under temperature and pressure controlled condition. Tetrahedron Lett. 2018, 59, 2075. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Wang, Z.; Fu, X. Visible light promoted hydration of alkynes catalyzed by rhodium (III) porphyrins. Chem. Commun. 2015, 51, 11896. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Jiang, D.; Li, S.; Shu, X.; Li, H.; Zhang, A.; Xu, J.; Ni, B. Visible light promoted copper-catalyzed Markovnikov hydration of alkynes at room temperature. Tetrahedron Lett. 2017, 58, 1156. [Google Scholar] [CrossRef]
- Mathur, P.; Joshi, R.K.; Jha, B.; Singh, A.K.; Mobin, S.M. Towards the catalytic formation of α, β-vinylesters and alkoxy substituted γ-lactones. J. Organomet. Chem. 2010, 695, 2687. [Google Scholar] [CrossRef]
- Mathur, P.; Joshi, R.K.; Rai, B.; Jha, D.K.; Mobin, S.M. One pot synthesis of maleimide and hydantoin by Fe(CO)5 catalyzed [2 + 2 + 1] co-cyclization of acetylene, isocyanate and CO. Dalton Trans. 2012, 41, 5045. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.; Jha, B.; Raghuvanshi, A.; Joshi, R.K.; Mobin, S.M. Photolytic reaction of substituted (ethynyl) benzaldehyde and Fe(CO)5: Formation of indenone and chelated iron complexes. J. Organomet. Chem. 2012, 712, 7. [Google Scholar] [CrossRef]
- Mathur, P.; Rai, D.K.; Tauqeer, M.; Joshi, R.K.; Lahiri, G.K.; Mobin, S.M. Synthesis, structure and redox property of first 1, 2, 3-triselenole. J. Organomet. Chem. 2012, 721, 144. [Google Scholar] [CrossRef]
- Jha, B.; Raghuvanshi, A.; Joshi, R.K.; Mobin, S.M.; Mathur, P. A photochemical route to ferrocenyl-substituted ferrapyrrolinone complexes. Appl. Organomet.Chem. 2017, 31, e3805. [Google Scholar] [CrossRef]
- Joshi, R.K.; Satrawala, N. One pot synthesis of important retinoid synthon by the catalytic regioselective bi-functionalization of acetylenes, alcohol and carbon monoxide. Tetrahedron Lett. 2017, 58, 2931. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Savelev, M.M. Metal carbonyl catalysts of the synthesis of organic compounds from carbon monoxide and molecular hydrogen. Russ. Chem. Rev. 1988, 57, 17. [Google Scholar] [CrossRef]
- Zhu, L.; Yempally, V.; Isrow, D.; Pellechia, P.J.; Captain, B. Selective benzylic C–H activation of solvent toluene and m-xylene by an iron–tin cluster complex: Fe2(μ-Sn )2(CO)8. J. Organomet. Chem. 2010, 695, 1. [Google Scholar] [CrossRef]
- Kaisare, A.A.; Jr, O.S.; Valente, E.J.; Gray, G.M. Metallacrown ethers with a symmetric bis(phosphite) ligand derived from 1,2-bis-(2-hydroxyethoxy)benzene: Synthesis, characterization and hydroformylation of styrene. J. Organomet. Chem. 2010, 695, 2658. [Google Scholar] [CrossRef]
- Tan, G.; Szilvási, T.; Inoue, S.; Blom, B.; Driess, M. An Elusive Hydridoaluminum(I) Complex for Facile C–H and C–O Bond Activation of Ethers and Access to Its Isolable Hydridogallium(I) Analogue: Syntheses, Structures, and Theoretical Studies. J. Am. Chem. Soc. 2014, 136, 9732. [Google Scholar] [CrossRef]
- Pandey, S.; Raj, K.V.; Shinde, D.R.; Vanka, K.; Kashyap, V.; Kurungot, S.; Vinod, C.P.; Chikkali, S.H. Iron catalyzed hydroformylation of alkenes under mild conditions: Evidence of an Fe (II) catalyzed process. J. Am. Chem. Soc. 2018, 140, 4430. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.F. Copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides. Commun. Chem. 2018, 1, 39. [Google Scholar] [CrossRef]
- Iwasaki, M.; Miki, N.; Ikemoto, Y.; Ura, Y.; Nishihara, Y. Regioselective Synthesis of γ-Lactones by Iron-Catalyzed Radical Annulation of Alkenes with α-Halocarboxylic Acids and Their Derivatives. Org. Lett. 2018, 20, 3848. [Google Scholar] [CrossRef]
- Xu, K.; Peng, H.; Lam, J.W.Y.; Poon, T.W.H.; Dong, Y.; Xu, H.; Sun, Q.; Cheuk, K.K.L.; Salhi, F.; Lee, P.P.S.; et al. Transition metal carbonyl catalysts for polymerizations of substituted acetylenes. Macromolecules 2000, 33, 6918. [Google Scholar] [CrossRef]
- Masuda, T.; Kuwane, Y.; Yamamoto, K.; Higashimura, T. Polymerization of acetylene derivatives induced by UV irradiation via metal carbonyls. Polym. Bull. 1980, 2, 823. [Google Scholar] [CrossRef]
- Landon, S.J.; Shulman, P.M.; Geoffrey, G.L. Photoassisted polymerization of terminal alkynes by W (CO) 6 involving catalyst generation by an alkyne to vinylidene ligand rearrangement. J. Am. Chem. Soc. 1985, 107, 6739. [Google Scholar] [CrossRef]
- Sharma, K.N.; Satrawala, N.; Srivastava, A.K.; Ali, M.; Joshi, R.K. Palladium (ii) ligated with a selenated (Se, C NHC, N−)-type pincer ligand: An efficient catalyst for Mizoroki–Heck and Suzuki–Miyaura coupling in water. Org. Biomol. Chem. 2019, 17, 8969. [Google Scholar] [CrossRef]
- Sharma, K.N.; Satrawala, N.; Joshi, R.K. Thioether–NHC-Ligated PdII Complex for Crafting a Filtration-Free Magnetically Retrievable Catalyst for Suzuki–Miyaura Coupling in Water. Eur. J. Inorg. Chem. 2018, 1743. [Google Scholar] [CrossRef]
- Sharma, C.; Srivastava, A.K.; Sharma, K.N.; Joshi, R.K. Half-sandwich (η5-Cp*)Rh(iii) complexes of pyrazolated organo-sulfur/selenium/tellurium ligands: Efficient catalysts for base/solvent free C–N coupling of chloroarenes under aerobic conditions. Org. Biomol. Chem. 2020, 18, 3599. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.N.; Ali, M.; Srivastava, A.K.; Joshi, R.K. (η6-Benzene) Ru (II) half-sandwich complexes of pyrazolated chalcogenoethers for catalytic activation of aldehydes to amides transformation. J. Organomet. Chem. 2019, 879, 69. [Google Scholar] [CrossRef]
- Yao, Q.; Kinney, E.P.; Zheng, C. Selenium-ligated palladium (ii) complexes as highly active catalysts for carbon−carbon coupling reactions: The heck reaction. Org. Lett. 2004, 6, 2997. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.I.M.; Lemke, F.R. Substituent effects in the ruthenium catalyzed hydrosilylation of para-substituted phenylacetylenes. Inorg. Chem. Commun. 2005, 8, 425. [Google Scholar] [CrossRef]
- Eckart, K.; Schwarz, H. Vinyl cation-induced cleavage of the oxygen-carbon bond in ortho-methoxy-substituted phenylacetylenes. J. Mass Spectrom. Ion Process. 1985, 66, 245. [Google Scholar] [CrossRef]
- Ramana, D.V.; Krishna, N.V.S.R. Ortho effects in organic molecules on electron impact: 18—Novel hydrogen transfer from the methoxy group to acetylenic carbon in 2-methoxyphenylacetylene and 2-methoxydiphenylacetylenes. J. Mass Spectrom. 1989, 24, 317. [Google Scholar]
- Dutta, A.K.; Maji, S.K.; Srivastava, D.N.; Mondal, A.; Biswas, P.; Paul, P.; Adhikary, B. Synthesis of FeS and FeSe Nanoparticles from a Single Source Precursor: A Study of Their Photocatalytic Activity, Peroxidase-Like Behavior, and Electrochemical Sensing of H2O2. ACS Appl. Mater. Interfaces 2012, 4, 1919. [Google Scholar] [CrossRef]
- Cong, B.; Sun, S.; Wang, B.; Lv, B.; Zhao, J.; Jin, F.; Jia, J.; Chen, G. Iron selenide nanoparticles-encapsulated within bamboo-like N-doped carbon nanotubes as composite anodes for superior lithium and sodium-ion storage. J. Chem. Eng. 2022, 435, 135185. [Google Scholar] [CrossRef]
- Hou, B.; Benito-Alifonso, D.; Webster, R.F.; Cherns, D.; Galan, M.C.; Fermín, D.J. Synthetic Mechanism Studies of Iron Selenides: An Emerging Class of Materials for Electrocatalysis. Catalysts 2021, 11, 681. [Google Scholar] [CrossRef]
- Oyetunde, T.; Omorogie, M.O.; O’Brien, P. Ferromagnetic FeSe2 from a mixed sulphur-selenium complex of iron [Fe{(SePPh2NPPh2S)2N}3] through pyrolysis. Heliyon 2020, 6, e03763. [Google Scholar] [CrossRef]
- Cho, J.S.; Park, J.; Kang, Y.C. Double-walled iron oxide nanotubes via selective chemical etching and Kirkendall process. Sci. Rep. 2020, 6, 38933. [Google Scholar] [CrossRef]
- Zheng, Q.; Cheng, X.; Li, H. Microwave Synthesis of High Activity FeSe2/C Catalyst toward Oxygen Reduction Reaction. Catalysts 2015, 5, 1079. [Google Scholar] [CrossRef]
- Hieber, W.; Gruber, J. Zur Kenntnis der Eisencarbonylchalkogenide. J. Inorg. Gen. Chem. 1958, 296, 91–103. [Google Scholar] [CrossRef]
- Polin, J.; Schottenberger, H. Conversion of methyl ketones into terminal acetylenes: Ethynylferrocene. Organic Syntheses 1998, 9, 411–414. [Google Scholar]
- Jacubert, M.; Provot, O.; Peyrat, J.F.; Hamze, A.; Brion, J.D.; Alami, M. p-Toluenesulfonic acid-promoted selective functionalization of unsymmetrical arylalkynes: A regioselective access to various arylketones and heterocycles. Tetrahedron 2010, 66, 3775–3787. [Google Scholar] [CrossRef]
- Nishizawa, M.; Skwarczynski, M.; Imagawa, H.; Sugihara, T. Mercuric triflate-TMU catalyzed hydration of terminal alkyne to give methyl ketone under mild conditions. Chem. Lett. 2002, 31, 12–13. [Google Scholar] [CrossRef]
- Jennings, P.W.; Hartman, J.W.; Hiscox, W.C. Alkyne hydration using Pt (II) catalysts. Inorg. Chim. Acta 1994, 222, 317–322. [Google Scholar] [CrossRef]
- Lumbroso, A.; Vautravers, N.R.; Breit, B. Rhodium-Catalyzed Selective anti-Markovnikov Addition of Carboxylic Acids to Alkynes. Org. Lett. 2010, 12, 5498–5501. [Google Scholar] [CrossRef]
- Park, Y.J.; Kwon, B.; Ahn, J.; Lee, H.; Jun, C. Chelation-assisted hydrative dimerization of 1-alkyne forming α, β-enones by an Rh (I) catalyst. J. Am. Chem. Soc. 2004, 126, 13892–13893. [Google Scholar] [CrossRef]
- Manikar, P.S.; Chippala, V.; Chegondi, R.; Chandrasekher, S. Ruthenium(II)-Catalyzed Hydration of Terminal Alkynes in PEG-400. Synlett 2016, 27, 1969–1972. [Google Scholar] [CrossRef]
- Kusakabe, T.; Ito, Y.; Kamimura, M.; Shirai, T.; Takahashi, K.; Mochida, T.; Kato, K. Palladium(II) Bis(oxazoline) Complexes that Catalyze the Hydration of Alkynes. Asian J. Org. Chem. 2017, 6, 1086–1090. [Google Scholar] [CrossRef]
- Marion, N.; Ramon, R.S.; Nolan, S.P. [(NHC)AuI]-Catalyzed Acid-Free Alkyne Hydration at Part-per-Million Catalyst Loadings. J. Am. Chem. Soc. 2009, 131, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Casado, R.; Contel, M.; Laguna, M.; Romero, P.; Sanz, S. Organometallic gold (III) compounds as catalysts for the addition of water and methanol to terminal alkynes. J. Am. Chem. Soc. 2003, 125, 11925–11935. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yeon, J.; Lee, P.H.; Lee, K. Iron-catalyzed indirect hydration of alkynes in presence of methanesulfonic acid. Tetrahedron Lett. 2013, 54, 4414–4417. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
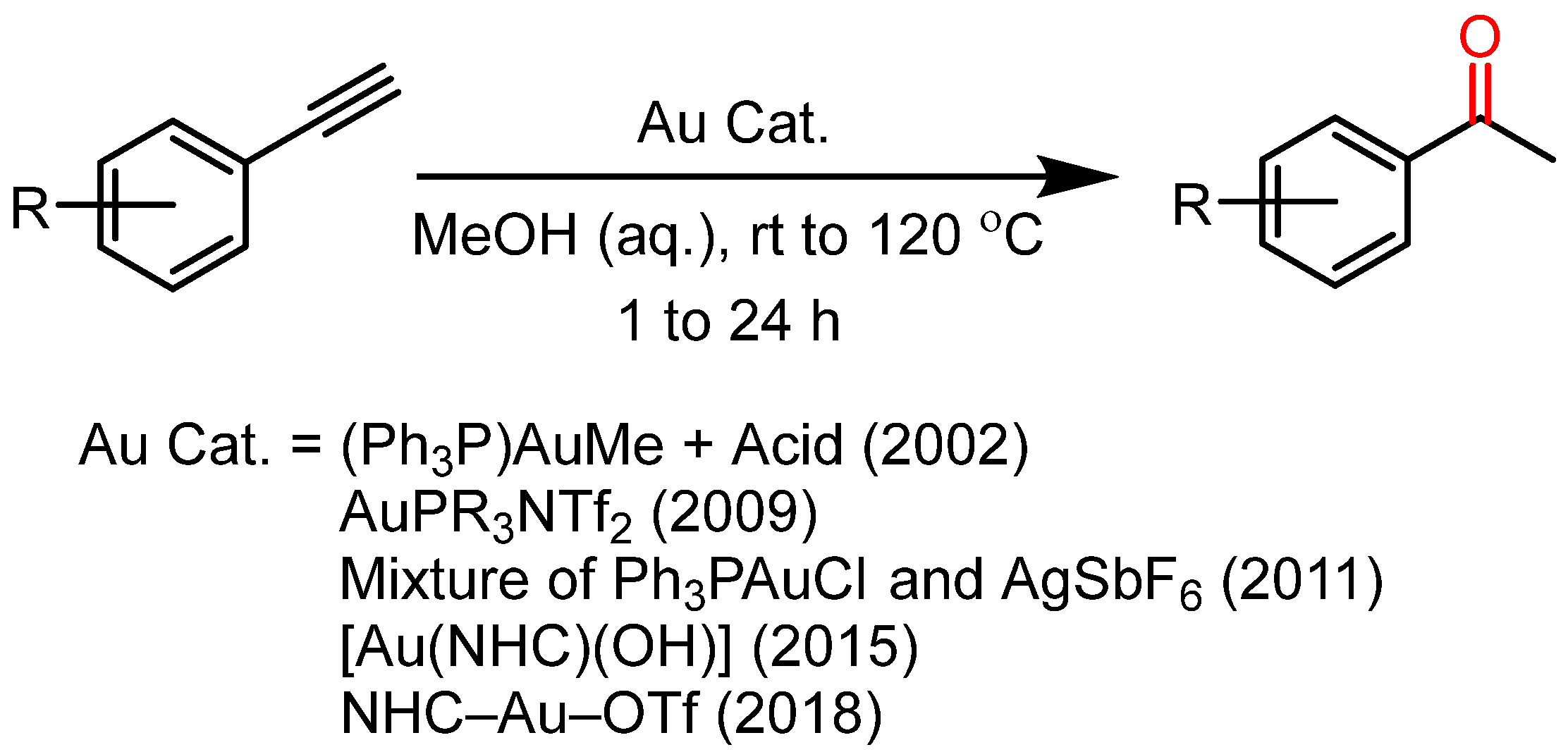{kind=link}
{kind=link}
{kind=link}
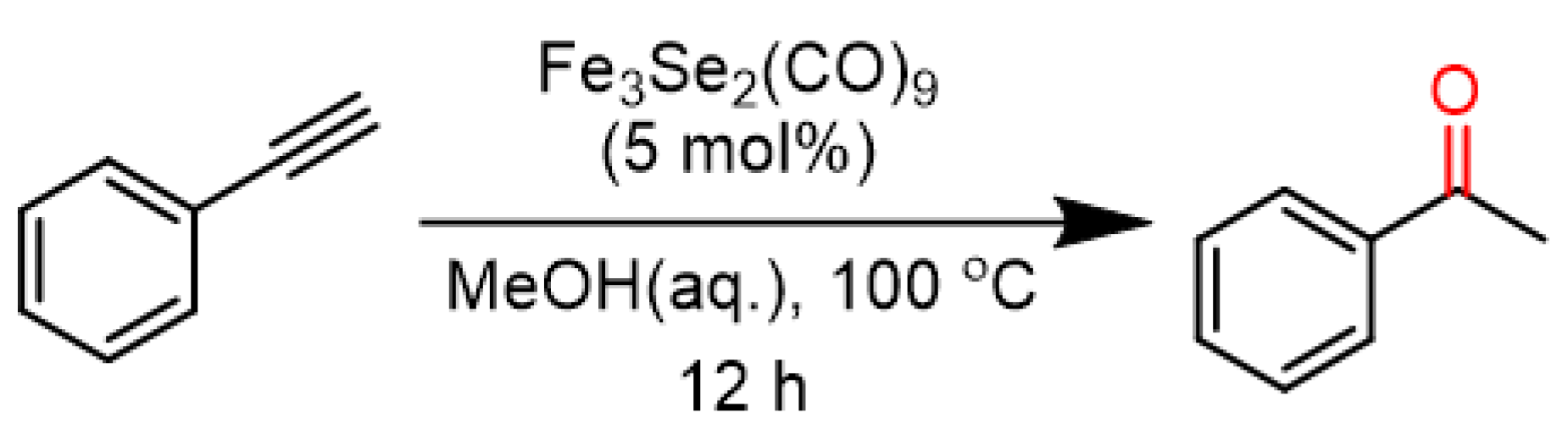{kind=link}

| Entry | Catalyst | Cat. Amt. (mol%) | Temp. (°C) | Time (min.) | Yield a (%) |
|---|---|---|---|---|---|
| 1. | - | - | −5 | 25 | - |
| 2. | Fe(CO)5 | 5 | −5 | 25 | - |
| 3. | Se | 5 | −5 | 25 | - |
| 4. | Fe2(CO)9 | 5 | −5 | 25 | - |
| 5. | Fe3(CO)12 | 5 | −5 | 25 | - |
| 6. | Fe3Se2(CO)9 | 5 | −5 | 25 | 86 |
| 7. | Fe3S2(CO)9 | 5 | −5 | 25 | 29 |
| 8. | Fe3Te2(CO)9 | 5 | −5 | 25 | 13 |
| 9. | Fe3Se2(CO)9 | 1 | −5 | 25 | - |
| 10. | Fe3Se2(CO)9 | 2 | −5 | 25 | 15 |
| 11. | Fe3Se2(CO)9 | 3 | −5 | 25 | 65 |
| 12. | Fe3Se2(CO)9 | 4 | −5 | 25 | 76 |
| 13. | Fe3Se2(CO)9 | 5 | −5 | 25 | 86 |
| 14. | Fe3Se2(CO)9 | 6 | −5 | 25 | 86 |
| 15. | Fe3Se2(CO)9 | 5 | 00 | 25 | 45 |
| 16. | Fe3Se2(CO)9 | 5 | −5 | 25 | 86 |
| 17. | Fe3Se2(CO)9 | 5 | −10 | 25 | 86 |
| 18. | Fe3Se2(CO)9 | 5 | −5 | 10 | 26 |
| 19. | Fe3Se2(CO)9 | 5 | −5 | 15 | 59 |
| 20. | Fe3Se2(CO)9 | 5 | −5 | 20 | 76 |
| 21. | Fe3Se2(CO)9 | 5 | −5 | 25 | 86 |
| 22. | Fe3Se2(CO)9 | 5 | −5 | 30 | 87 |
| Entry | Reactant | Product | Yield a (%) |
|---|---|---|---|
| 1a |  |  | 86 |
| 1b |  |  | 89 |
| 1c |  |  | 84 |
| 1d |  |  | 82 |
| 1e |  |  | 85 |
| 1f |  |  | 86 |
| 1g |  |  | 87 |
| 1h |  |  | 84 |
| 1i |  |  | 86 |
| 1j |  |  | 80 |
| 1k |  |  | 74 |
| 1l |  |  | 75 |
| 1m |  |  | 86 |
| 1n |  |  | 85 |
| 1o |  |  | 80 |
| 1p |  |  | 78 |
| 1q |  |  | 82 |
| Entry | Reactant | Product | Yield a (%) |
|---|---|---|---|
| 2a |  |  | 71 |
| 2b |  |  | 70 |
| 2c |  |  | 73 |
| 2d |  |  | 65 |
| 2e |  |  | 68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.; Srivastava, A.K.; Upadhyay, N.S.; Satrawala, N.; Joshi, R.K. Facile Photochemical/Thermal Assisted Hydration of Alkynes Catalysed under Aqueous Media by a Chalcogen Stabilized, Robust, Economical, and Reusable Fe3Se2(CO)9 Cluster. Organics 2023, 4, 251-264. https://doi.org/10.3390/org4020020
Ali M, Srivastava AK, Upadhyay NS, Satrawala N, Joshi RK. Facile Photochemical/Thermal Assisted Hydration of Alkynes Catalysed under Aqueous Media by a Chalcogen Stabilized, Robust, Economical, and Reusable Fe3Se2(CO)9 Cluster. Organics. 2023; 4(2):251-264. https://doi.org/10.3390/org4020020
Chicago/Turabian StyleAli, Munsaf, Avinash K. Srivastava, Nitinkumar Satyadev Upadhyay, Naveen Satrawala, and Raj K. Joshi. 2023. "Facile Photochemical/Thermal Assisted Hydration of Alkynes Catalysed under Aqueous Media by a Chalcogen Stabilized, Robust, Economical, and Reusable Fe3Se2(CO)9 Cluster" Organics 4, no. 2: 251-264. https://doi.org/10.3390/org4020020