
Regioselective N- versus P-Deprotonation of Aminophosphane Tungsten(0) Complexes

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
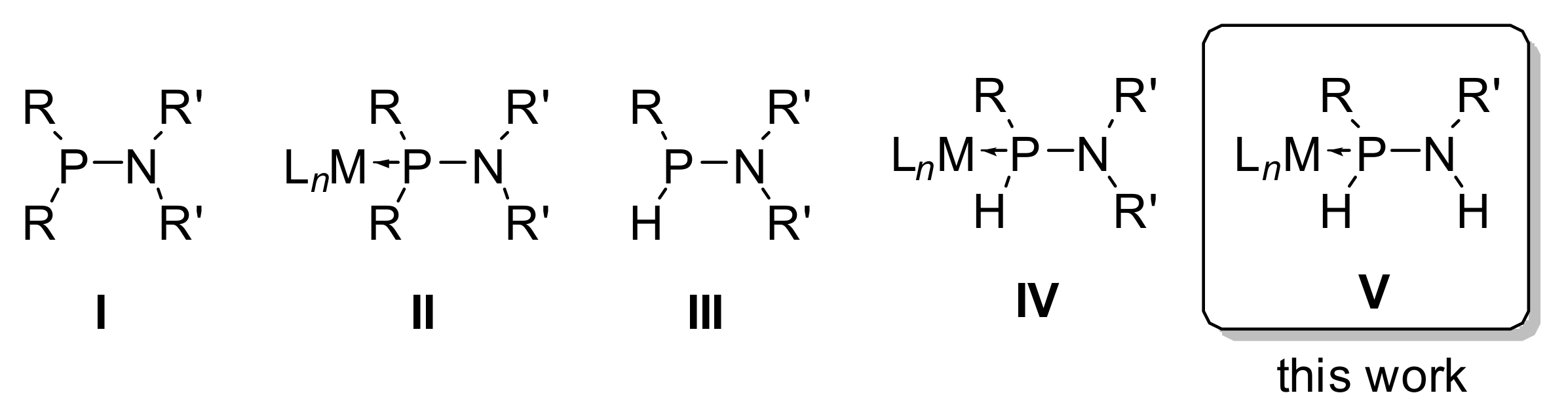
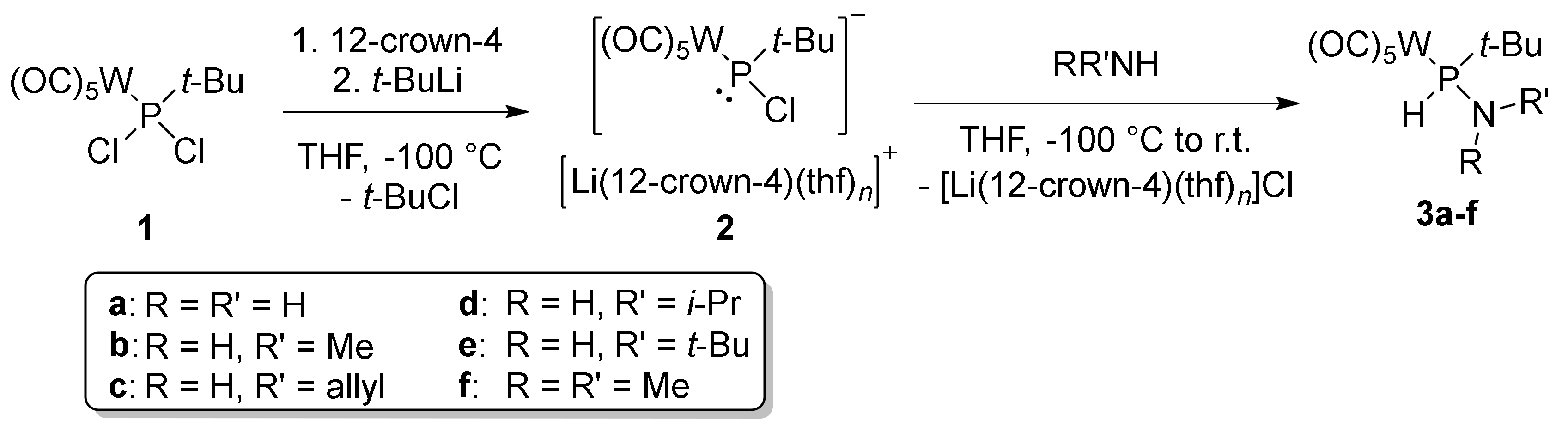
3.1. Synthesis and Characterization of P–tert–Bu Substituted Aminophosphane Complexes
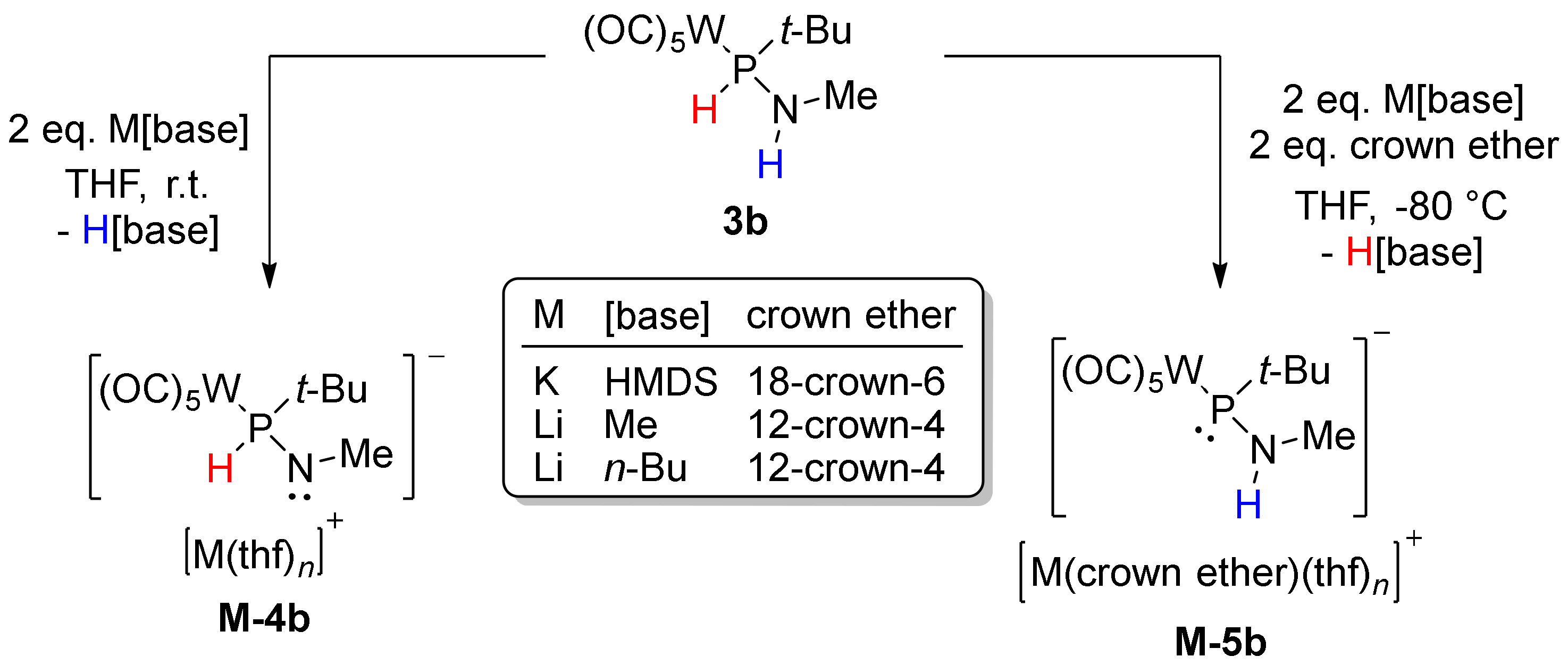
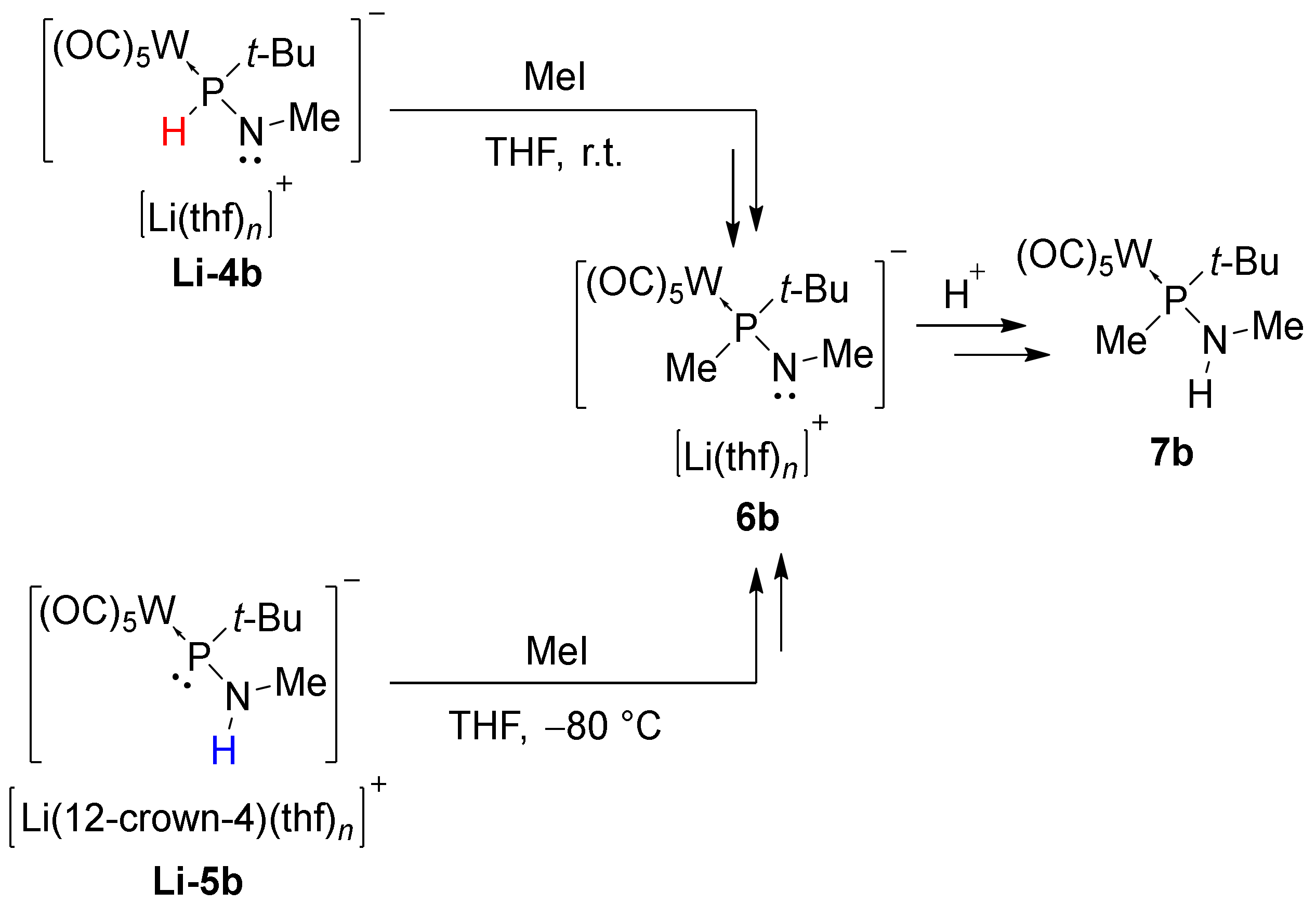
3.2. Regioselective Deprotonation of 1,2-Bifunctional Aminophosphane Complexes
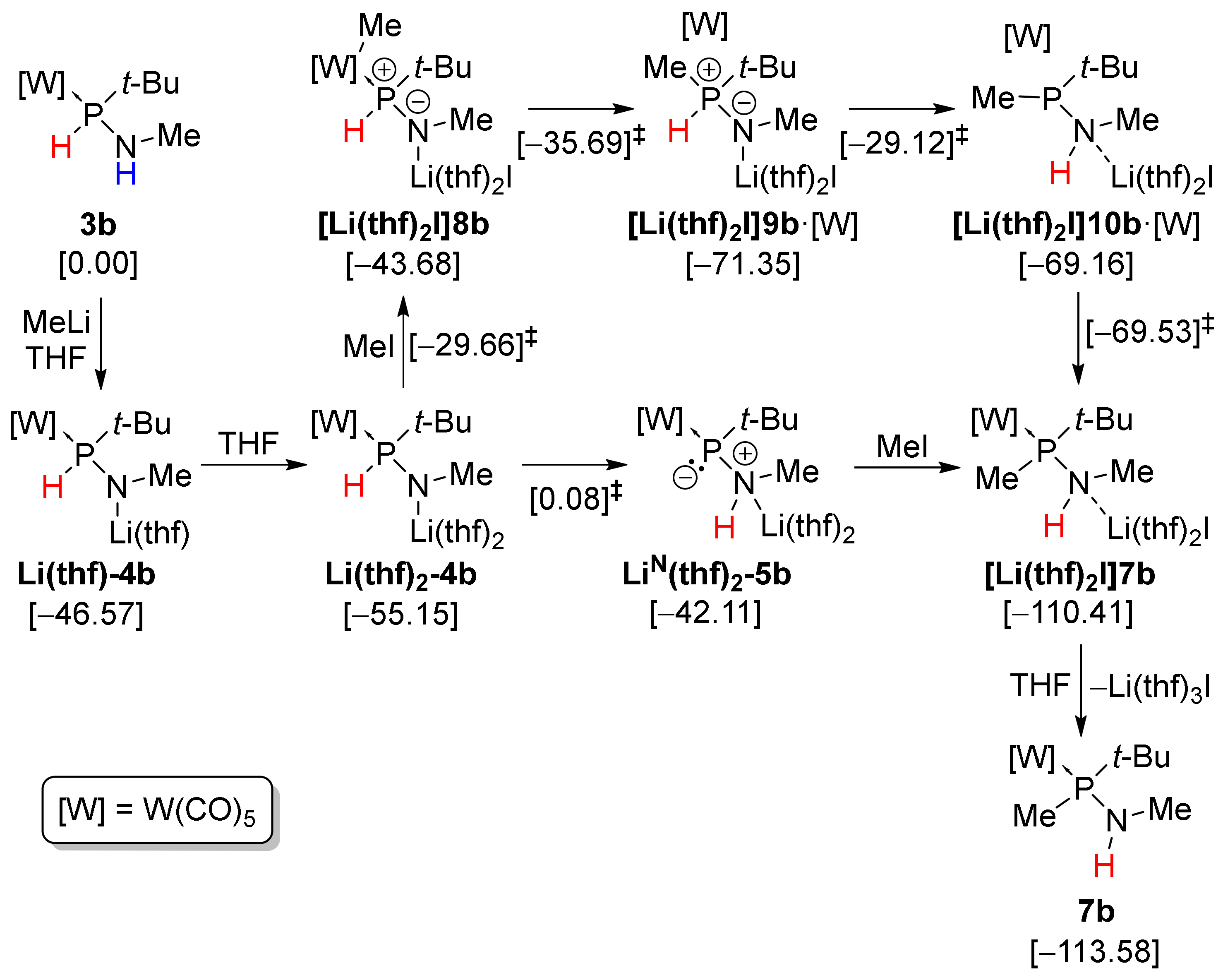
3.3. Theoretical Investigations on the Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, G.S. The Reaction of Chloro(bistrifluoromethyl)phosphine with Amines and Ammonia. Proc. Chem. Soc. 1957, 105–128, 118–119. [Google Scholar] [CrossRef]
- Burg, A.B.; Slota, P.J. Dimethylaminodimethylphosphine. J. Am. Chem. Soc. 1958, 80, 1107–1109. [Google Scholar] [CrossRef]
- Sisler, H.; Smith, N. Some N-Substituted Aminodiphenylphosphines. J. Org. Chem. 1961, 26, 611–613. [Google Scholar] [CrossRef]
- Atkinson, L.K.; Smith, D.C. Molybdenum carbonyl complexes of (dialkylamino)-diphenylphosphines (R2NPPh2). J. Organomet. Chem. 1971, 33, 189–194. [Google Scholar] [CrossRef]
- Kroshefsky, R.D.; Verkade, J.G.; Pipal, J.R. Coordination properties of constrained aminophosphanes. Phosphorus Sulfur Relat. Elem. 1979, 6, 377–389. [Google Scholar] [CrossRef]
- Huttner, G.; Müller, H.-D. Stabilization of Unknown Phosphanes: Secondary Amino- and Alkoxyphosphanes as Complex Ligands. Angew. Chem. Int. Ed. Engl. 1975, 14, 571–572. [Google Scholar] [CrossRef]
- Marinetti, A.; Mathey, F. Stabilization of R-P(H)A species (A = OH, OR, S, NH2, NHR, NR2, Cl, Br, I) by complexation with chromium and tungsten pentacarbonyls. Organometallics 1982, 1, 1488–1492. [Google Scholar] [CrossRef]
- Mathey, F. Phospha-Organic Chemistry: Panorama and Perspectives. Angew. Chem. Int. Ed. Engl. 2003, 42, 1578–1604. [Google Scholar] [CrossRef]
- Wit, J.B.M.; de Jong, G.B.; Schakel, M.; Lutz, M.; Ehlers, A.W.; Slootweg, J.C.; Lammertsma, K. iPr2N–P=Fe(CO)4 in Olefinic Solvents: A Reservoir of a Transient Phosphinidene Complex Capable of Substrate Hopping. Organometallics 2016, 35, 1170–1176. [Google Scholar] [CrossRef]
- Niecke, E.; Ringel, G. Synthesis of a secondary aminophosphine. Angew. Chem. Int. Ed. Engl. 1977, 16, 486–487, Erratum in Angew. Chem. 1977, 89, 501–502. [Google Scholar] [CrossRef]
- Niecke, E.; Rüger, R. Bis(trimethylsilyl)aminophosphane. Angew. Chem. Int. Ed. Engl. 1982, 21, 62–71. [Google Scholar] [CrossRef]
- Duan, L.; Schnakenburg, G.; Daniels, J.; Streubel, R. P-OR functional phosphanido and/or Li/OR phosphinidenoid complexes? Eur. J. Inorg. Chem. 2012, 2012, 3490–3499. [Google Scholar] [CrossRef]
- Junker, P.; Qu, Z.-W.; Kalisch, T.; Schnakenburg, G.; Espinosa Ferao, A.; Streubel, R. A case study on the conversion of Li/Cl phosphinidenoid into phosphinidene complexes. Dalton Trans. 2021, 50, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Schmer, A.; Volk, N.; Espinosa Ferao, A.; Streubel, R. Access and unprecedented reaction pathways of Li/Cl phosphinidenoid iron(0) complexes. Dalton Trans. 2019, 48, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Majhi, P.K.; Kyri, A.W.; Schmer, A.; Schnakenburg, G.; Streubel, R. Synthesis and deprotonation of aminophosphane complexes: Potassium phosphinidenoid complexes and a new synthetic protocol for complexes with three-membered P-ring ligands. Chem. Eur. J. 2016, 22, 15413–15419. [Google Scholar] [CrossRef]
- Streubel, R.; Schmer, A.; Kyri, A.W.; Schnakenburg, G. 1,1′-Bifunctional Aminophosphane Complexes via N–H Bond Insertions of a Li/Cl Phosphinidenoid Complex and First Studies on N/P Mono Functionalizations. Organometallics 2017, 36, 1488–1495. [Google Scholar] [CrossRef]
- Schmer, A.; Terschüren, T.; Schnakenburg, G.; Espinosa Ferao, A.; Streubel, R. Access to 1,1′-Bifunctional Phosphane Iron(0) Complexes via P-N Bond-Forming Reactions and Selective P-Functionalizations. Eur. J. Inorg. Chem. 2019, 2019, 1604–1611. [Google Scholar] [CrossRef]
- Schmer, A.; Junker, P.; Espinosa Ferao, A.; Streubel, R. M/X Phosphinidenoid Metal Complex Chemistry. Acc. Chem. Res. 2021, 54, 1754–1765. [Google Scholar] [CrossRef]
- Streubel, R.; Kyri, A.W.; Duan, L.; Schnakenburg, G. Synthesis of Li/OR phosphinidenoid complexes: On the evidence for intramolecular O–Li donation and the effect of cation encapsulation. Dalton Trans. 2014, 43, 2088–2097. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Marinetti, A.; Bauer, S.; Ricard, L.; Mathey, F. The “Phospha-Wittig” Reaction: A New Method for Building Phosphorus Carbon Double and Single Bonds from Carbonyl Compounds. Organometallics 1990, 9, 793–798. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Deglmann, P.; May, K.; Furche, F.; Ahlrichs, R. Nuclear second analytical derivative calculations using auxiliary basis set expansions. Chem. Phys. Lett. 2004, 384, 103–107. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [Google Scholar] [CrossRef] [PubMed]
- Schreckenbach, G.; Ziegler, T. Calculation of NMR Shielding Tensors Using Gauge-Including Atomic Orbitals and Modern Density Functional Theory. J. Phys. Chem. 1995, 99, 606–611. [Google Scholar] [CrossRef]
- Fassbender, J.; Schnakenburg, G.; Espinosa Ferao, A.; Streubel, R. Effects of diminished steric protection at phosphorus on stability and reactivity of oxaphosphirane complexes. Dalton Trans. 2018, 47, 9347–9354. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.E.; Mingos, D.M.P. Determination of the Tolman cone angle from parameters and a statistical analysis using the Data Base. Transit. Met. Chem. 1995, 20, 533–539. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | R’ | δ(31P)/ppm | 1JW,P/Hz | 1JP,H/Hz | yield/% | |
|---|---|---|---|---|---|---|
| 3a | H | H | 45.7 | 239.0 | 333.5 | 56 |
| 3b | H | Me | 67.3 | 238.2 | 337.0 | 69 |
| 3c | H | Allyl | 61.6 | 239.0 | 339.4 | 80 |
| 3d | H | i-Pr | 55.5 | 238.5 | 334.0 | 69 |
| 3e | H | t-Bu | 37.1 | 239.9 | 340.6 | - |
| 3f | Me | Me | 91.0 | 239.5 | 358.8 | - |
| δ(31P)/ppm | δ(31P)calc/ppm[a] | 1JW,P/Hz | 1JP,H/Hz | |
|---|---|---|---|---|
| 3b | 67.3 | 67.3 | 238.2 | 337.0 |
| 3f | 91.0 | 86.9 | 239.5 | 358.8 |
| Li-4b | 49.6 | 43.6 (4b−) 67.9 (Li-4b) [b] 69.5 (thf-Li-4b) [b] 72.6 (Li-12c4-4b) [b] | 189.0 | 310.4 |
| K-4b | 48.5 | 52.3 (K-4b) [b] 62.4 (K-18c6-4b) [b] | 188.8 | 309 |
| Li-5b | 89.9 (br) | 95.2 (5b−) 87.3 (thf-Li-12c4-5b) [b] 68.1 (Li-12c4-5b) [b] | - [c] | - |
| K-5b | 89.1 | 86.3 (K-5b) [b] 77.1 (K-18c6-5b) [b] | 76.1 | - |
| Li-6b | 55.0 | 78.3 (6b−) 72.6 (Li-12c4-6b) [b] | 196.5 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terschüren, T.; Junker, P.; Schmer, A.; Espinosa Ferao, A.; Streubel, R. Regioselective N- versus P-Deprotonation of Aminophosphane Tungsten(0) Complexes. Organics 2022, 3, 161-172. https://doi.org/10.3390/org3030013
Terschüren T, Junker P, Schmer A, Espinosa Ferao A, Streubel R. Regioselective N- versus P-Deprotonation of Aminophosphane Tungsten(0) Complexes. Organics. 2022; 3(3):161-172. https://doi.org/10.3390/org3030013
Chicago/Turabian StyleTerschüren, Tatjana, Philip Junker, Alexander Schmer, Arturo Espinosa Ferao, and Rainer Streubel. 2022. "Regioselective N- versus P-Deprotonation of Aminophosphane Tungsten(0) Complexes" Organics 3, no. 3: 161-172. https://doi.org/10.3390/org3030013