
13C NMR Spectroscopic Studies of Intra- and Intermolecular Interactions of Amino Acid Derivatives and Peptide Derivatives in Solutions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
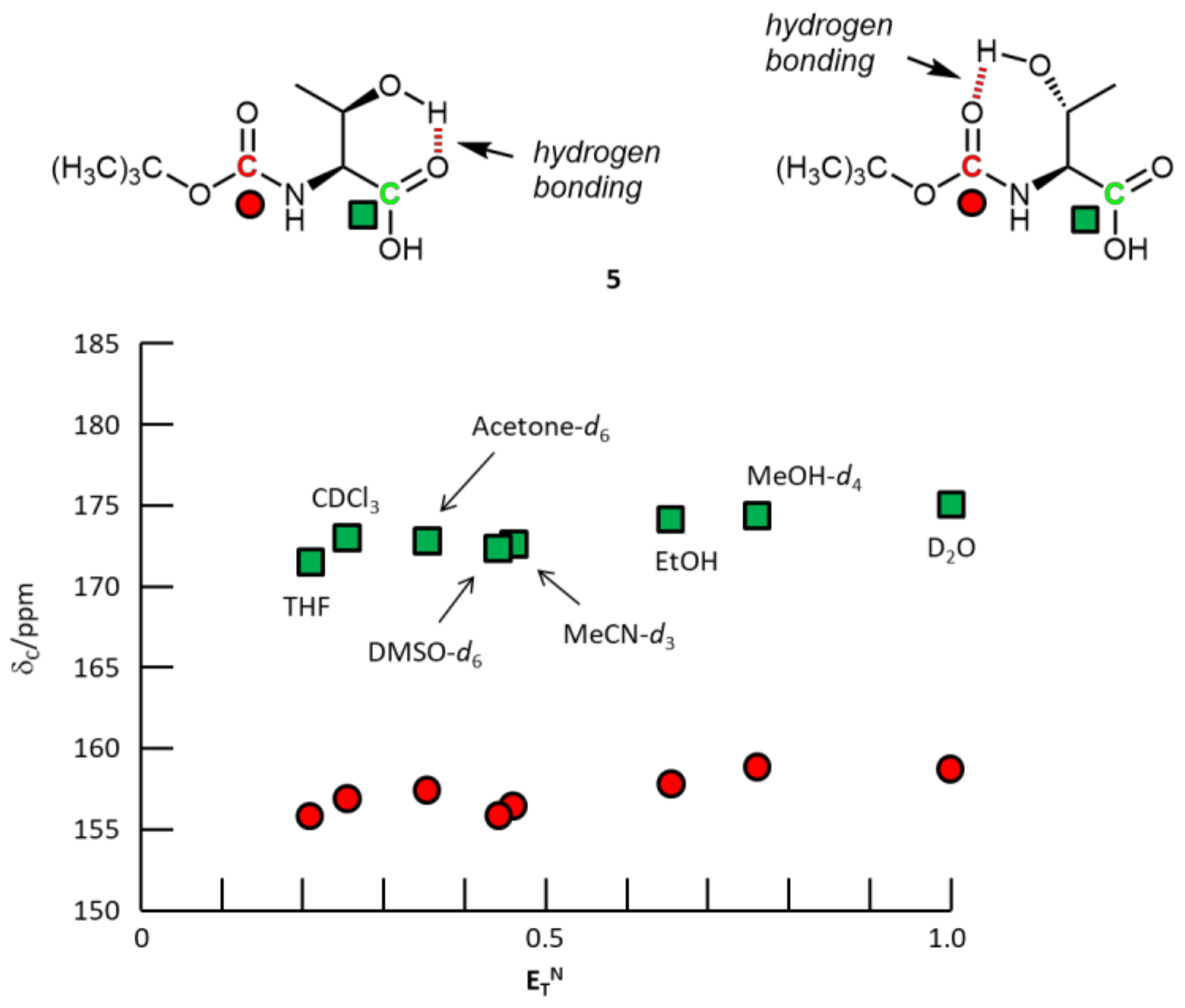{kind=link}
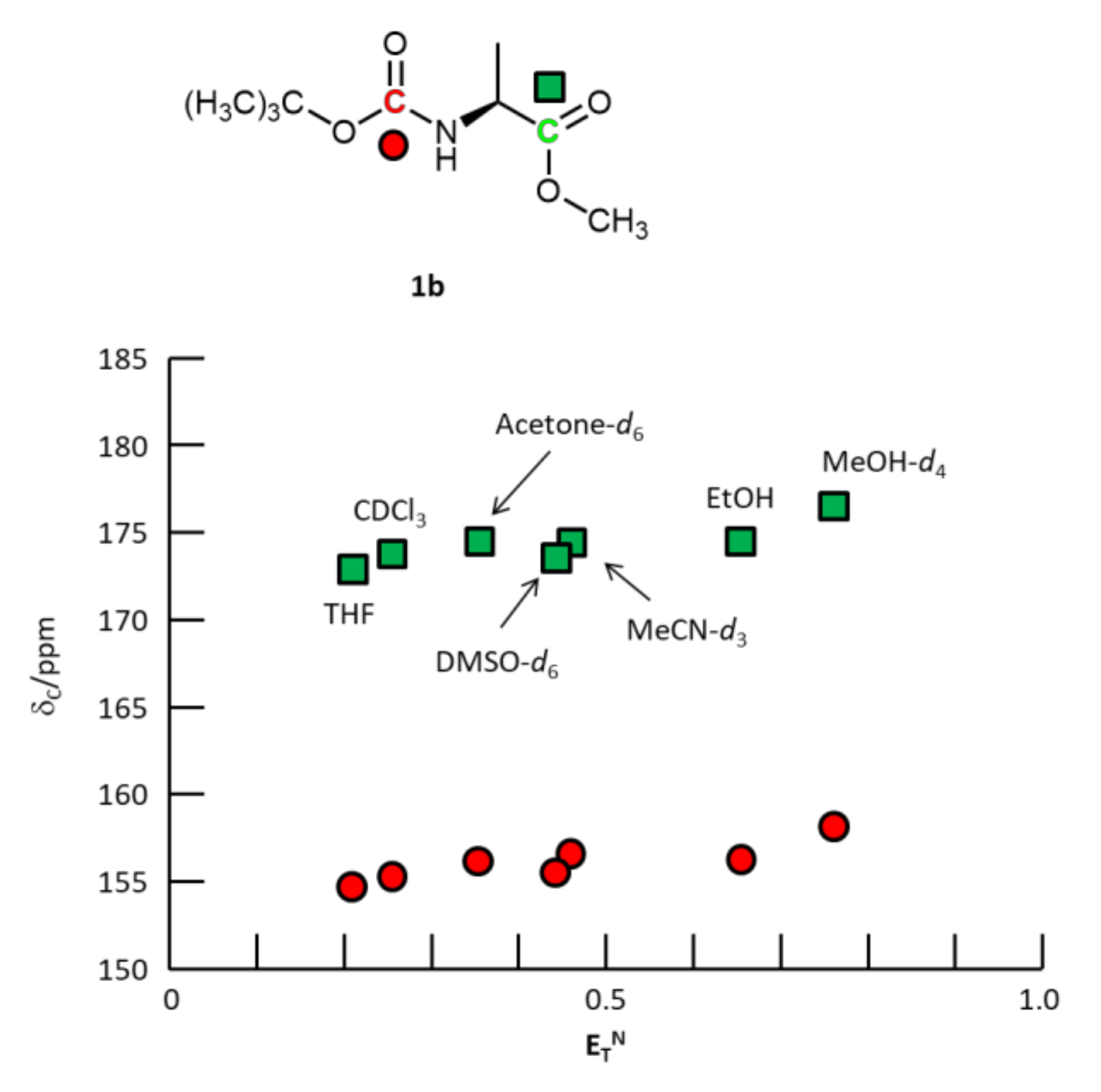{kind=link}
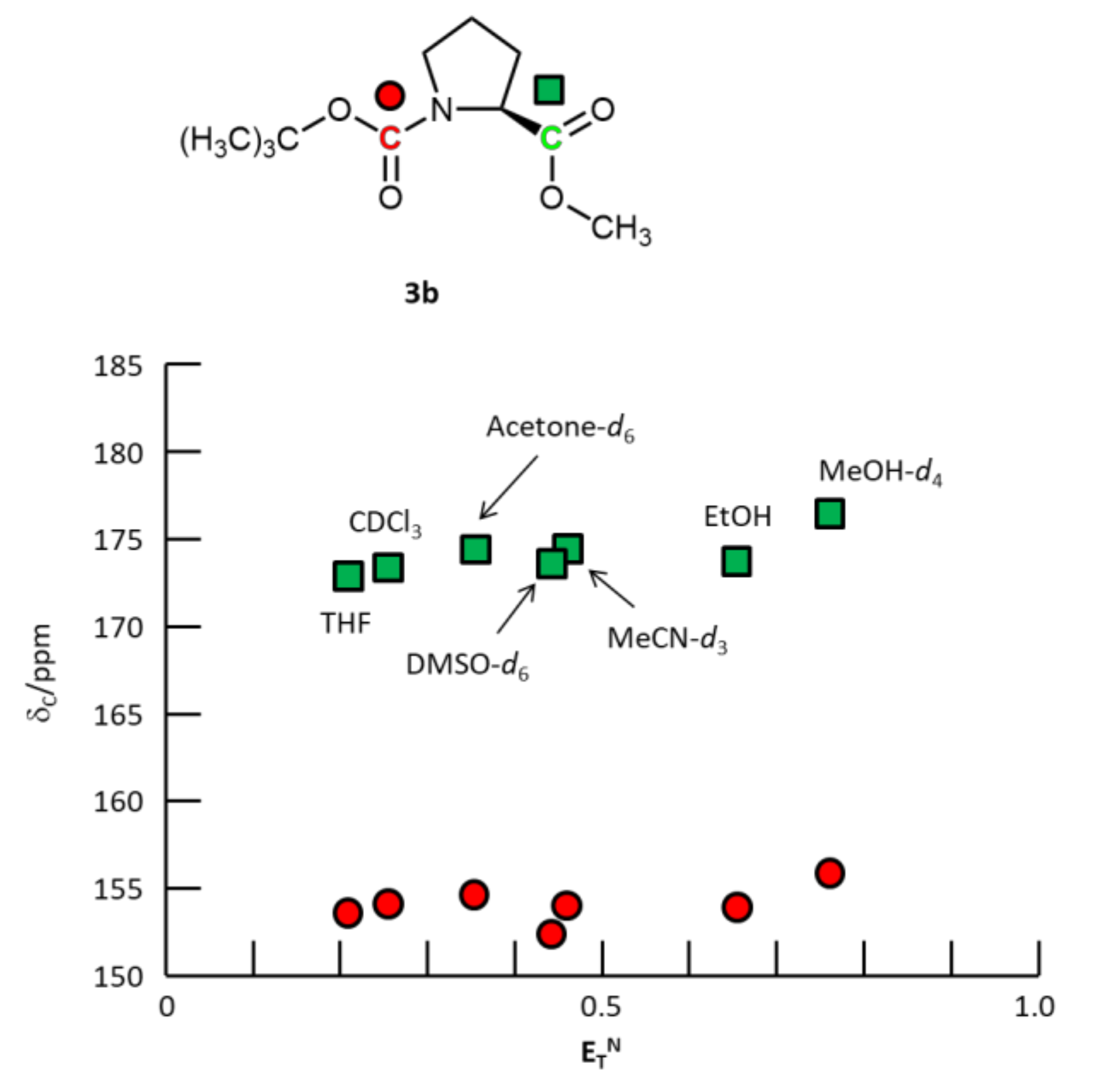{kind=link}
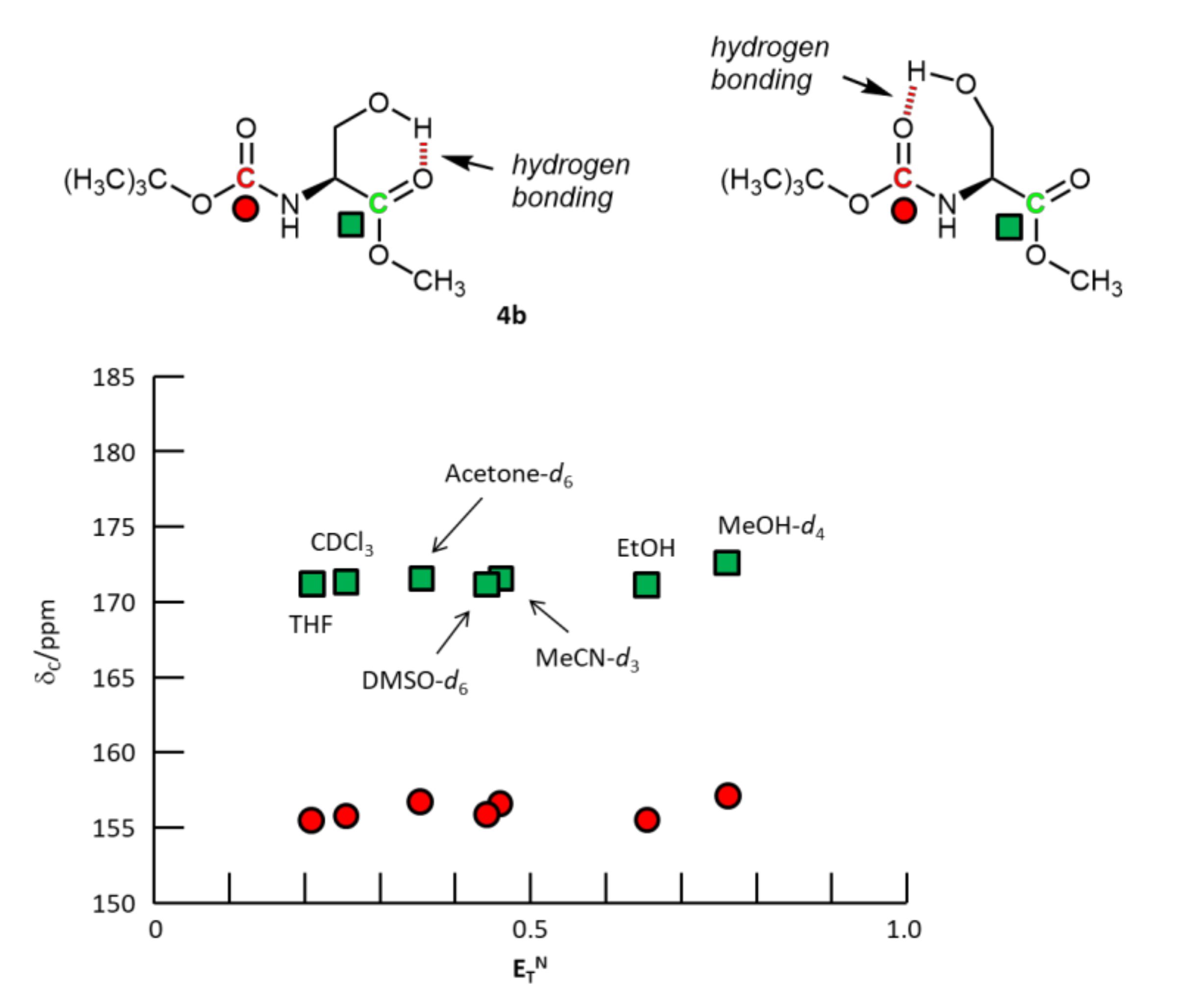{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. 13C NMR Measurements of Various Amino Acid Derivatives and Dipeptide Derivatives in Various Solvents
2.2. General Procedure for Methylation of N-Boc-Protected Amino Acids
2.2.1. N-Boc-L-alanine-OMe 1b
2.2.2. N-Boc-L-proline-OMe 3b
2.2.3. N-Boc-L-serine-OMe 4b
2.3. General Procedure for Preparation of N-Boc-Protected Dipeptide Derivatives
2.3.1. N-Boc-L-proline-L-alanine-OMe 6
2.3.2. N-Boc-L-proline-L-serine-OMe 7
2.3.3. N-Boc-L-proline-L-proline-OMe 8
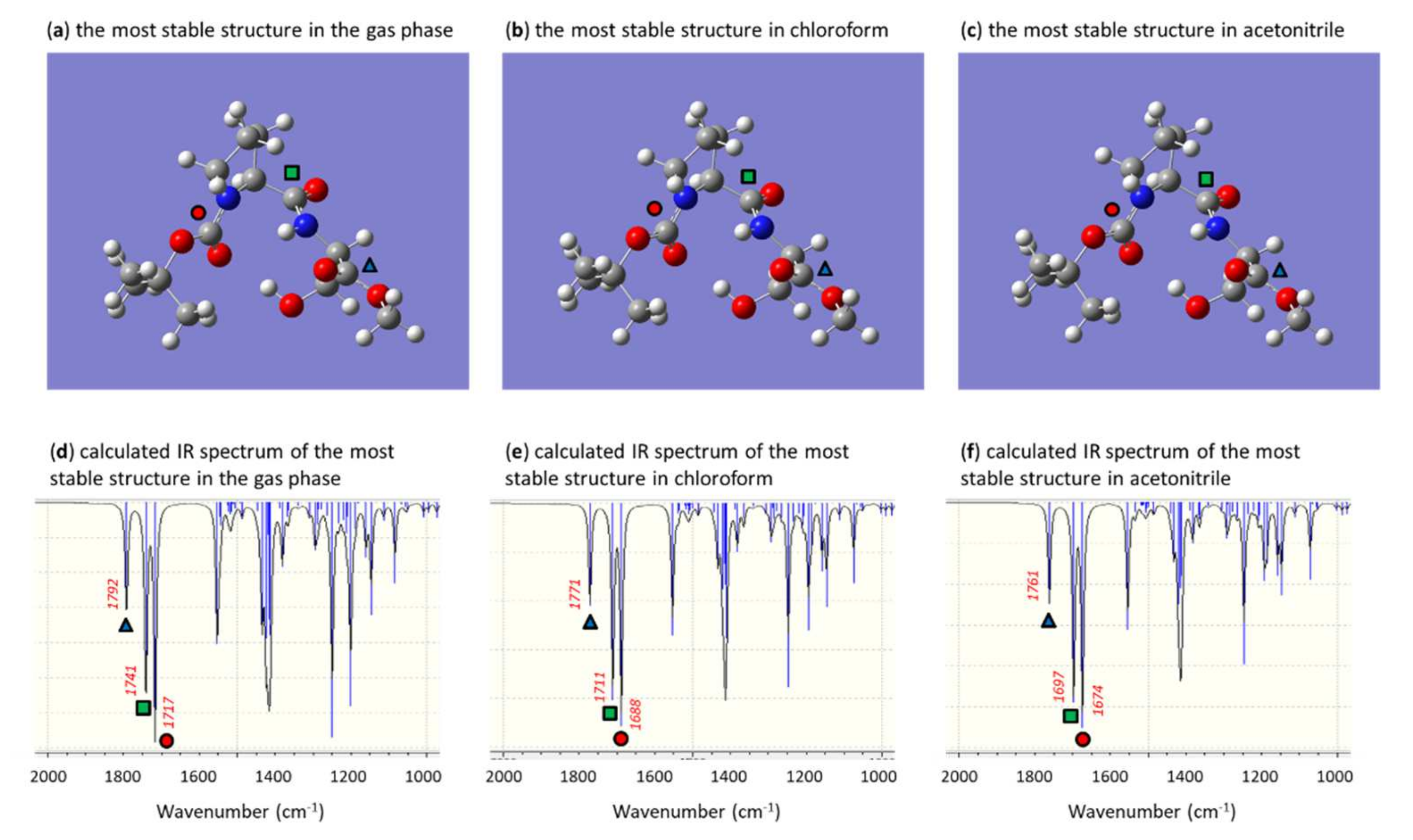
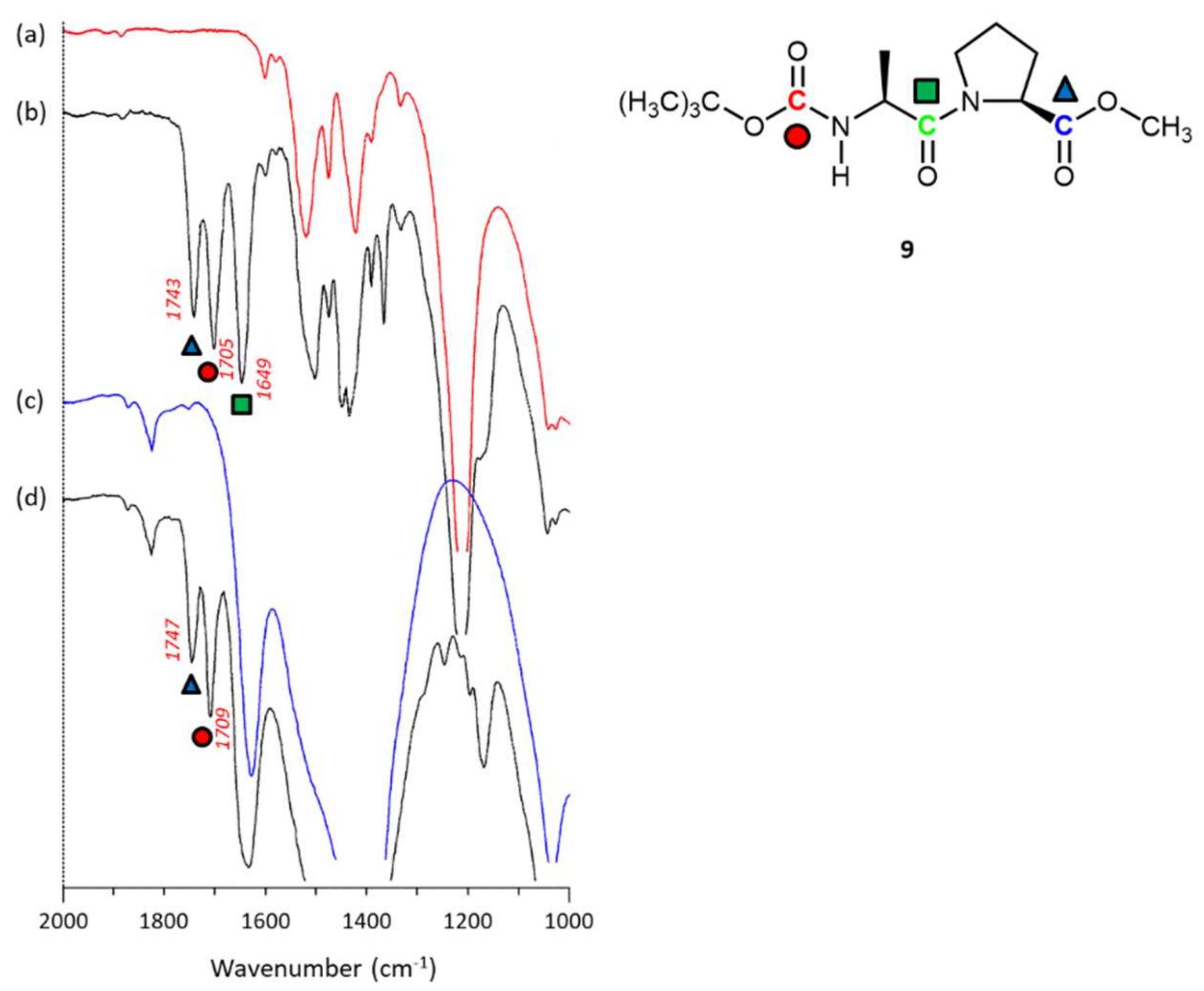
2.3.4. N-Boc-alanine-L-proline-OMe 9
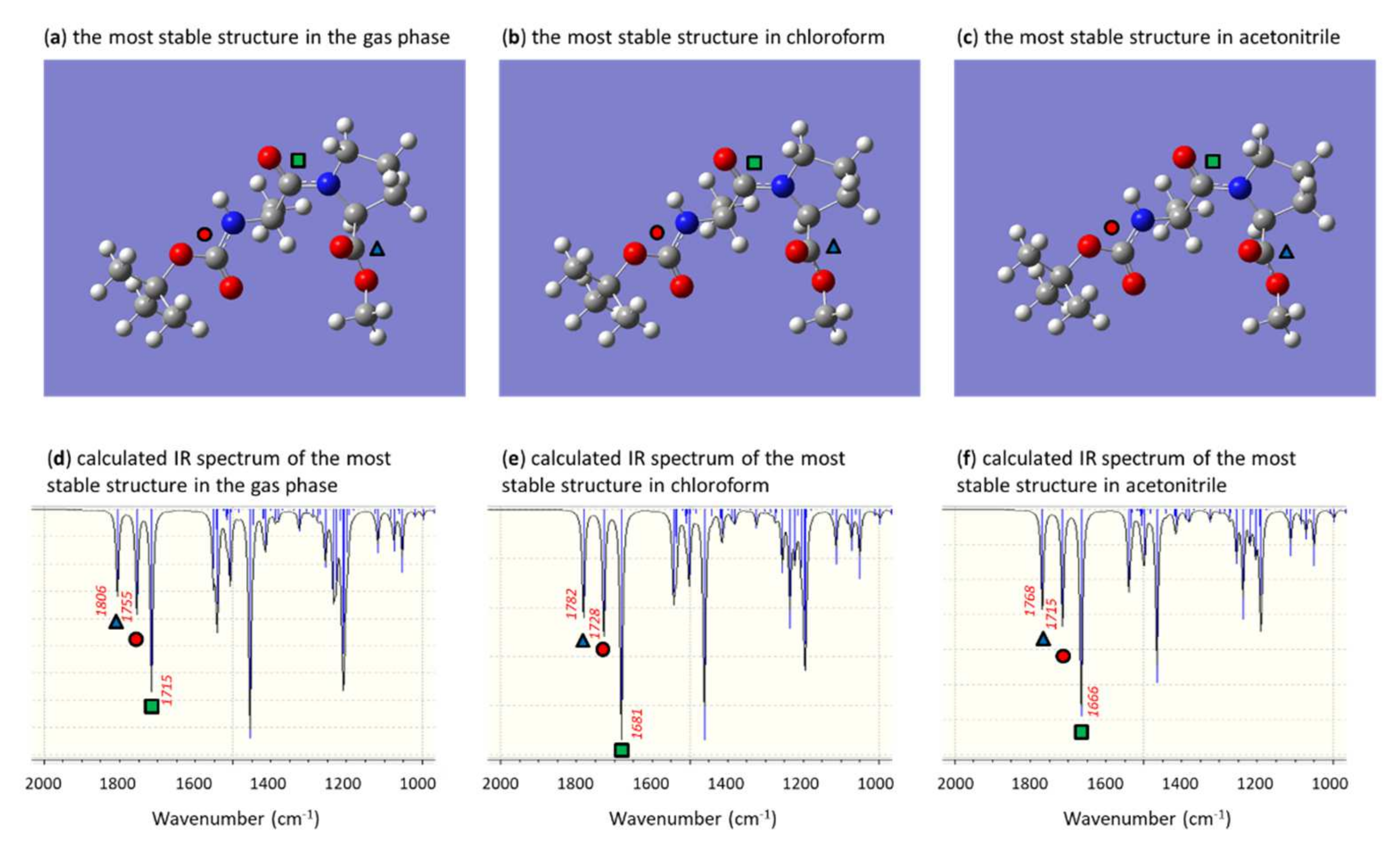
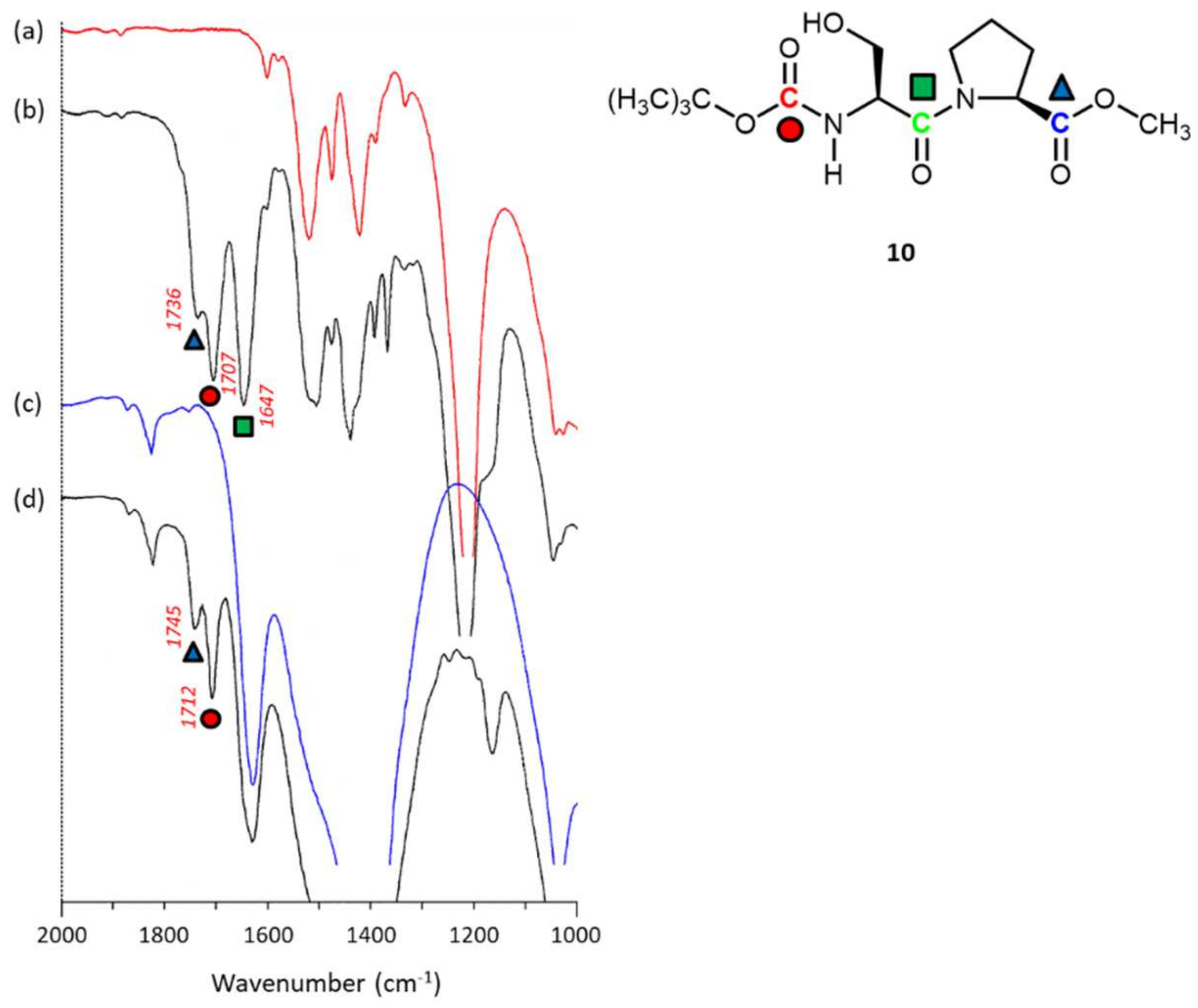
2.3.5. N-Boc-L-serine-L-proline-COOMe 10
2.4. Computational Methods
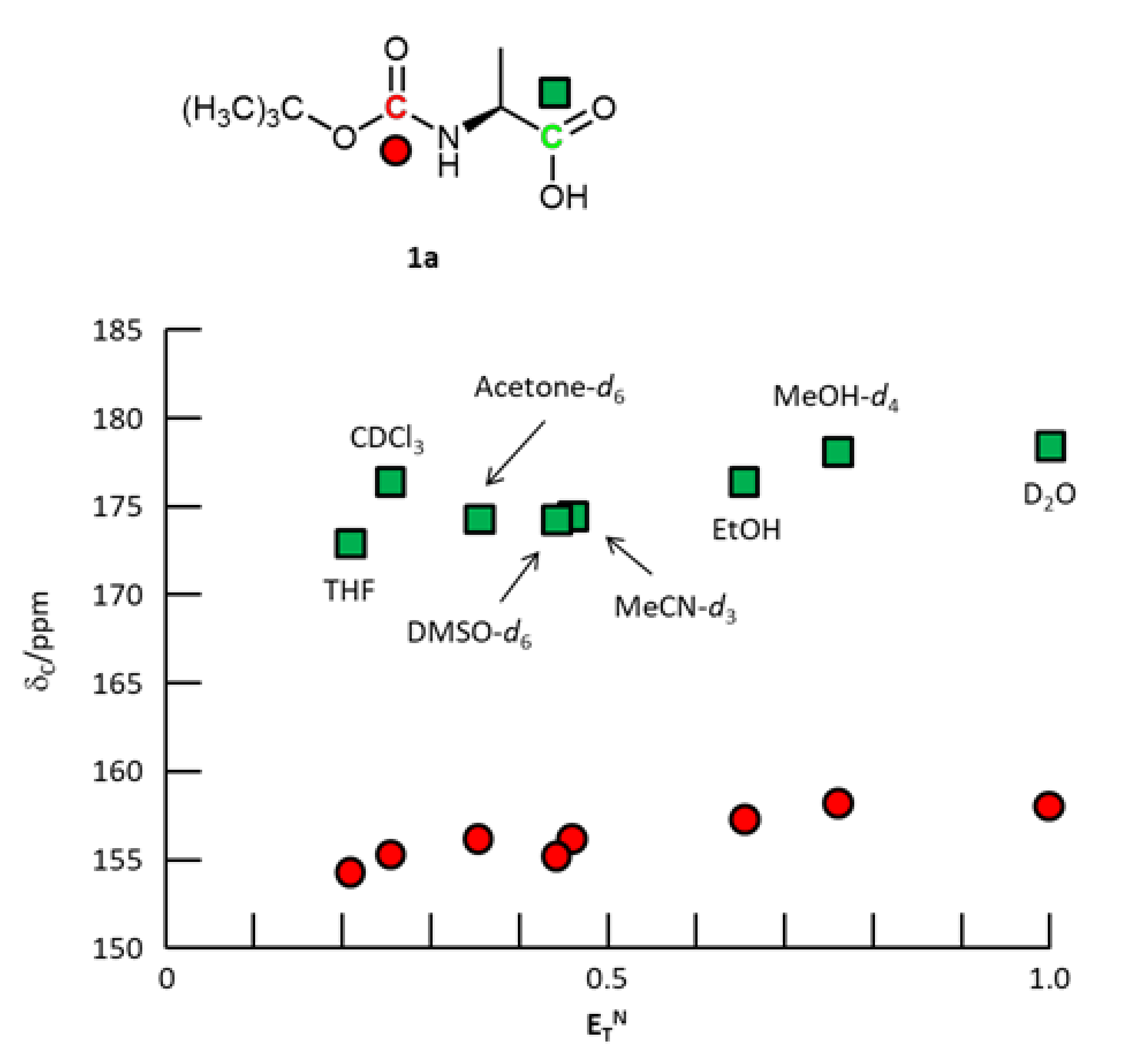
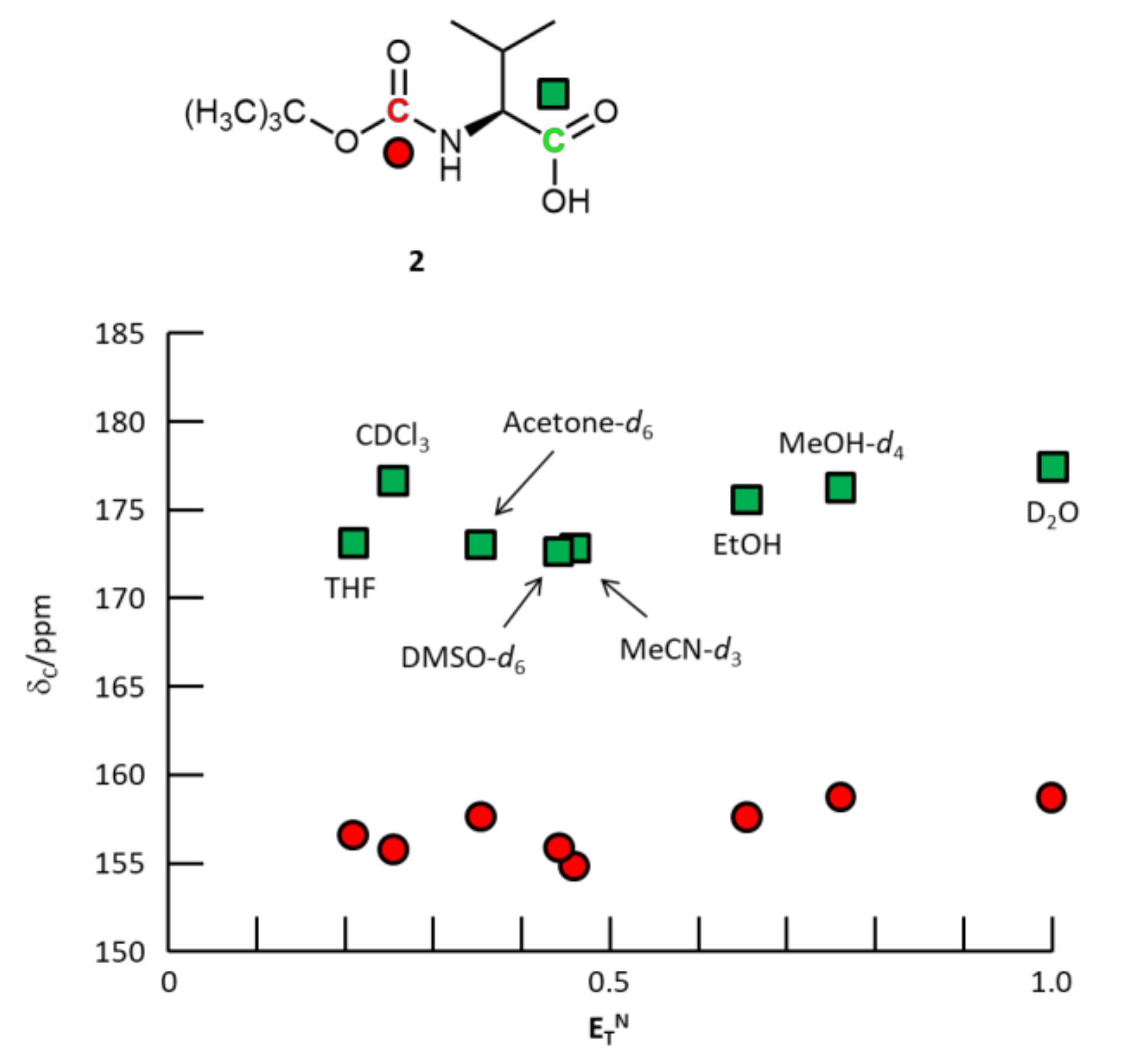
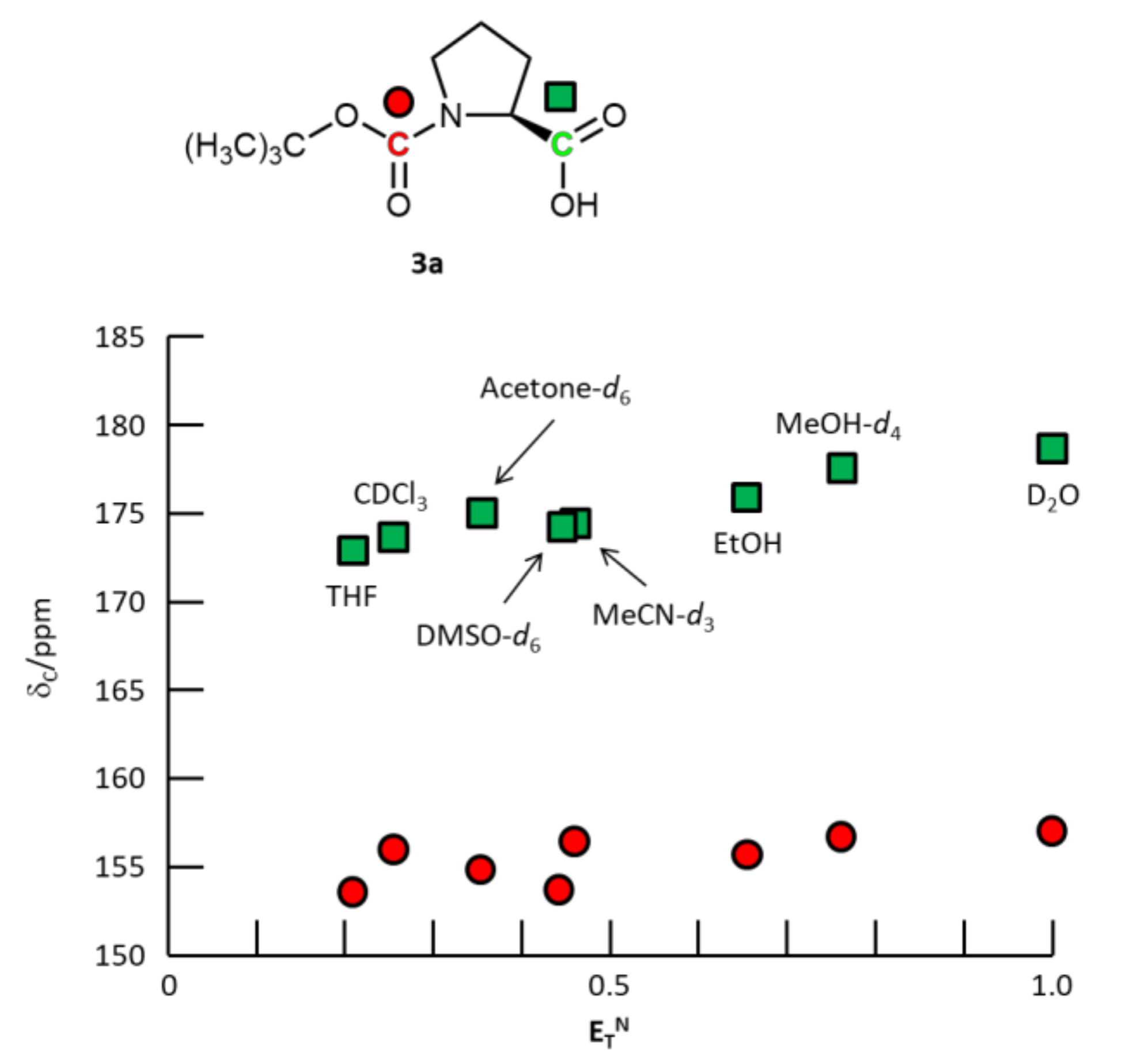
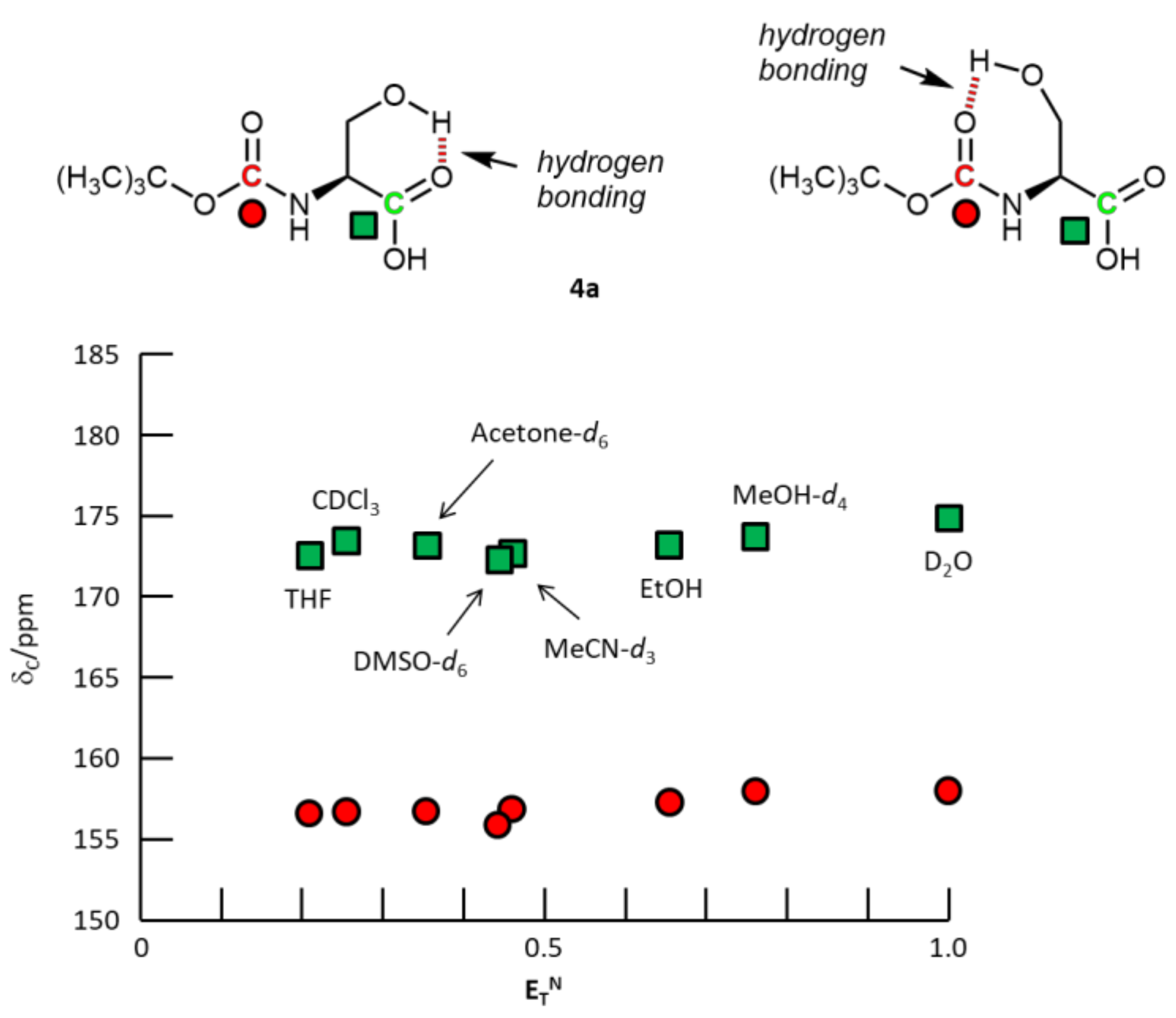
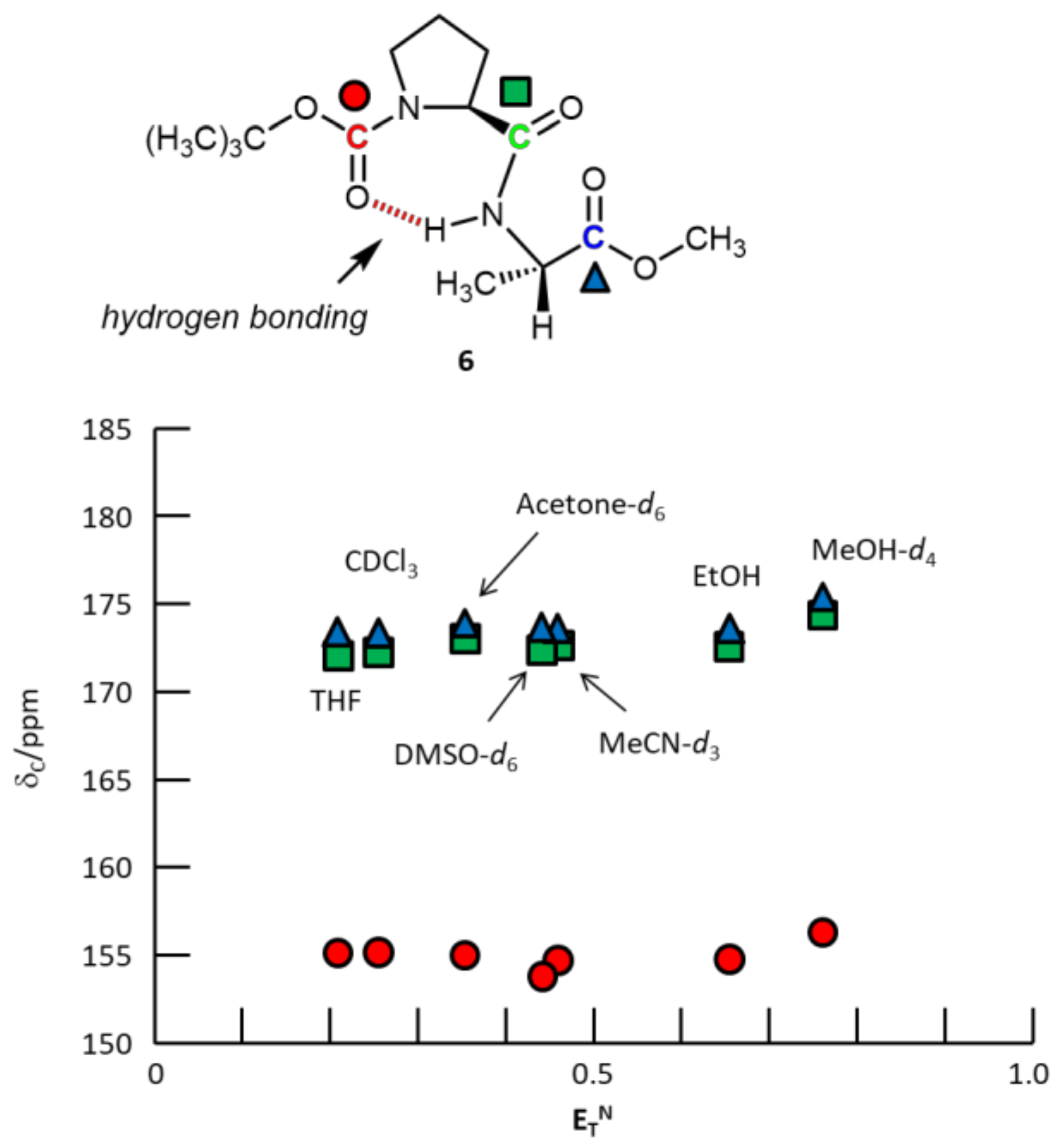
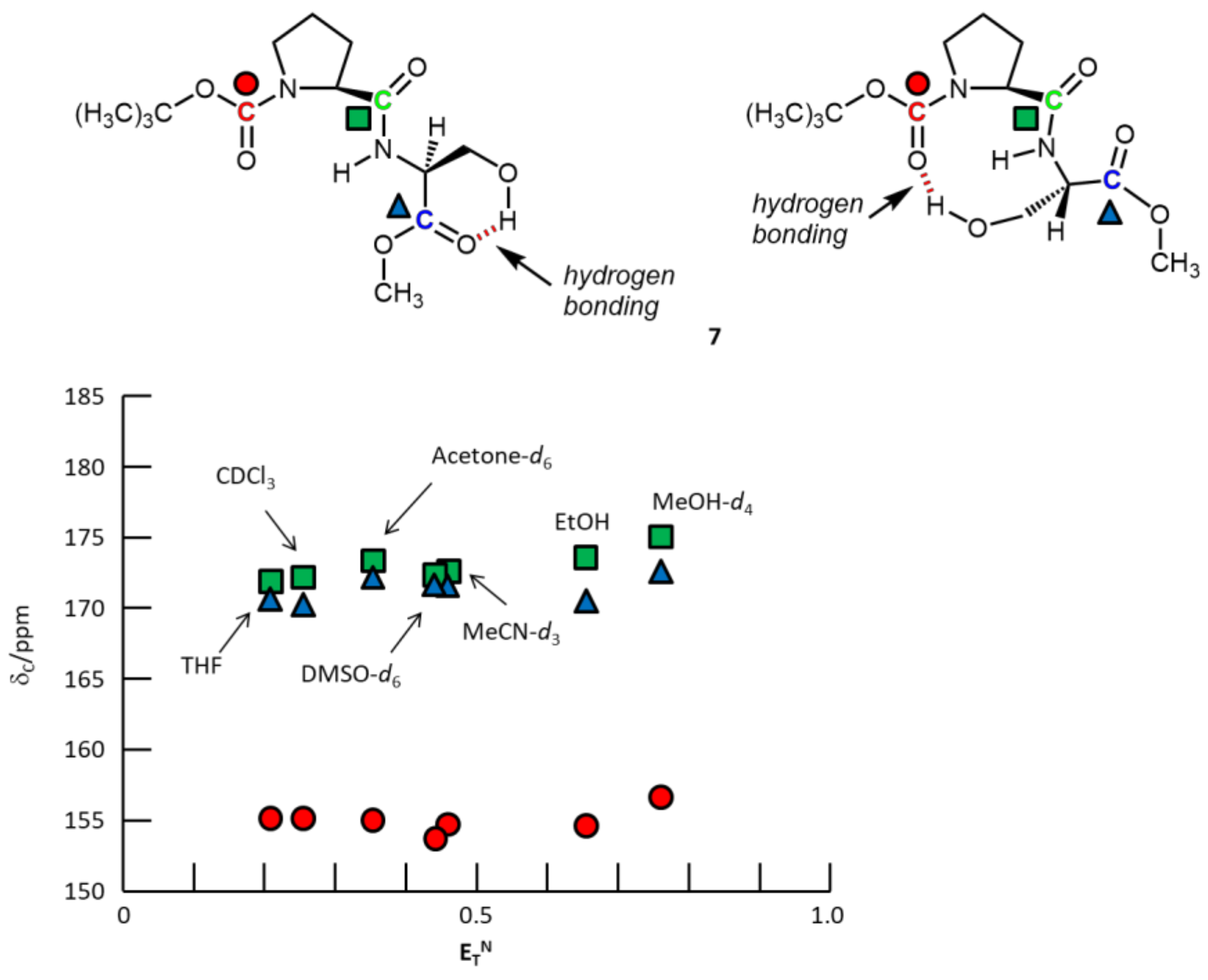
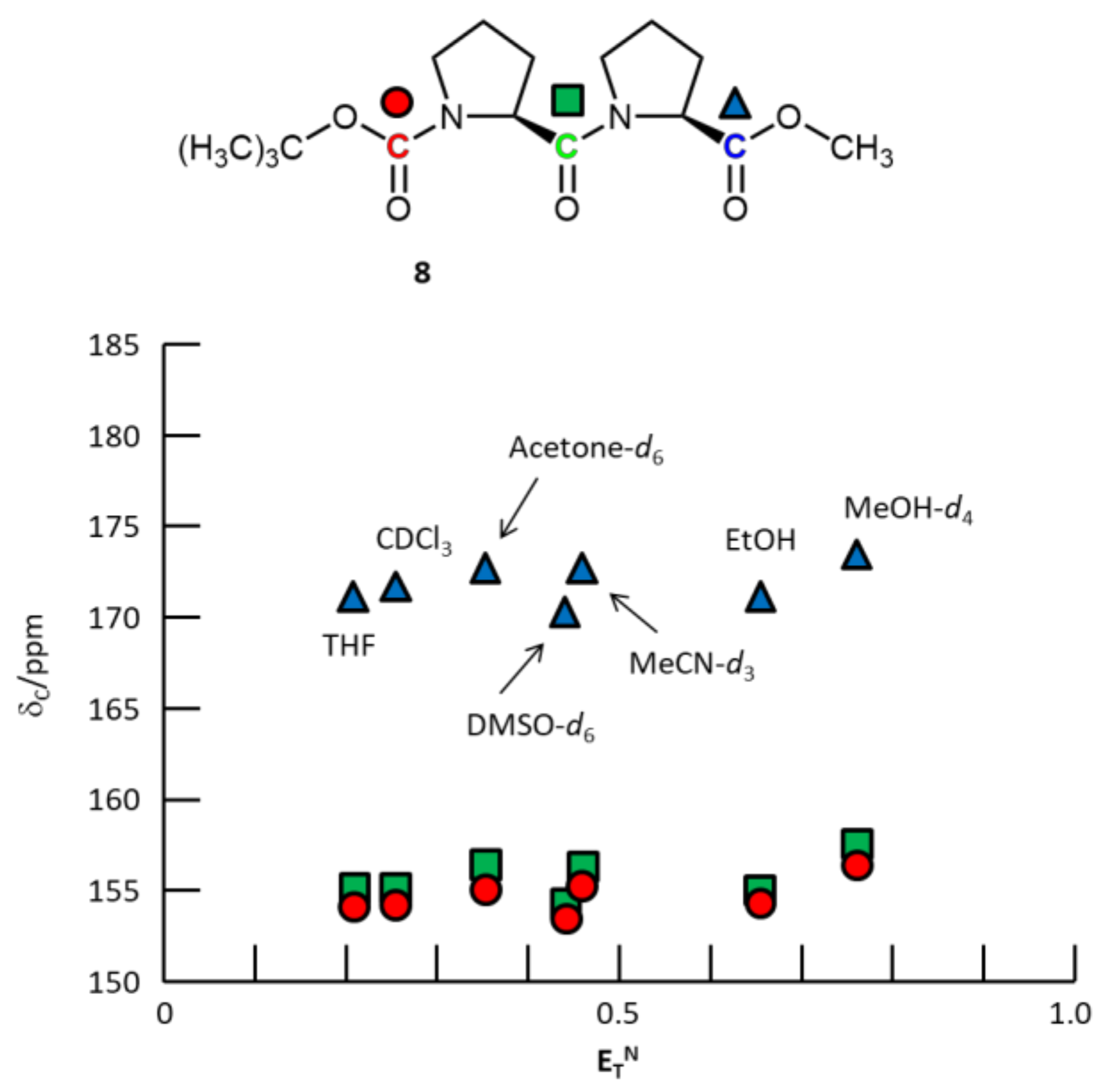
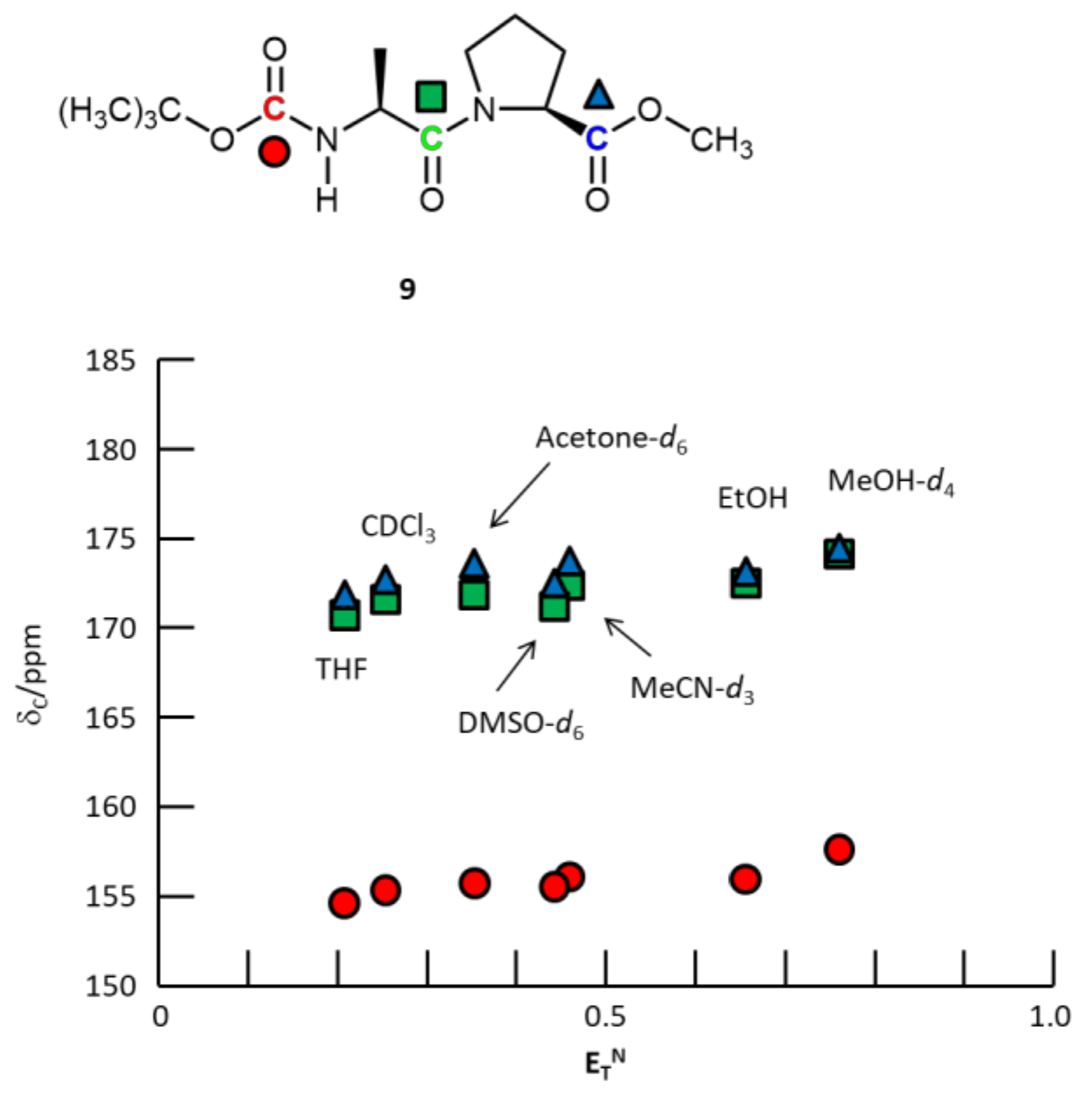
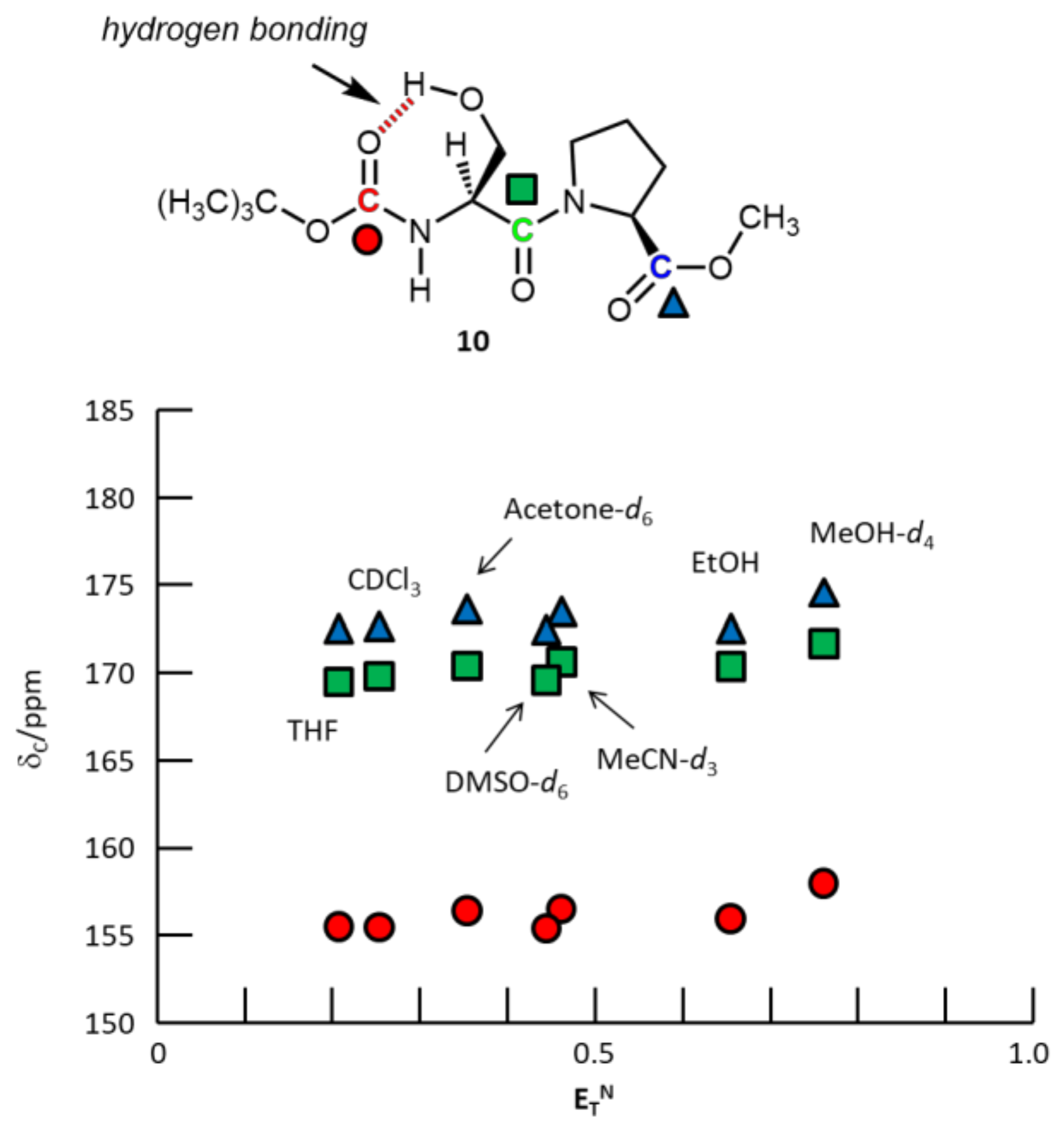
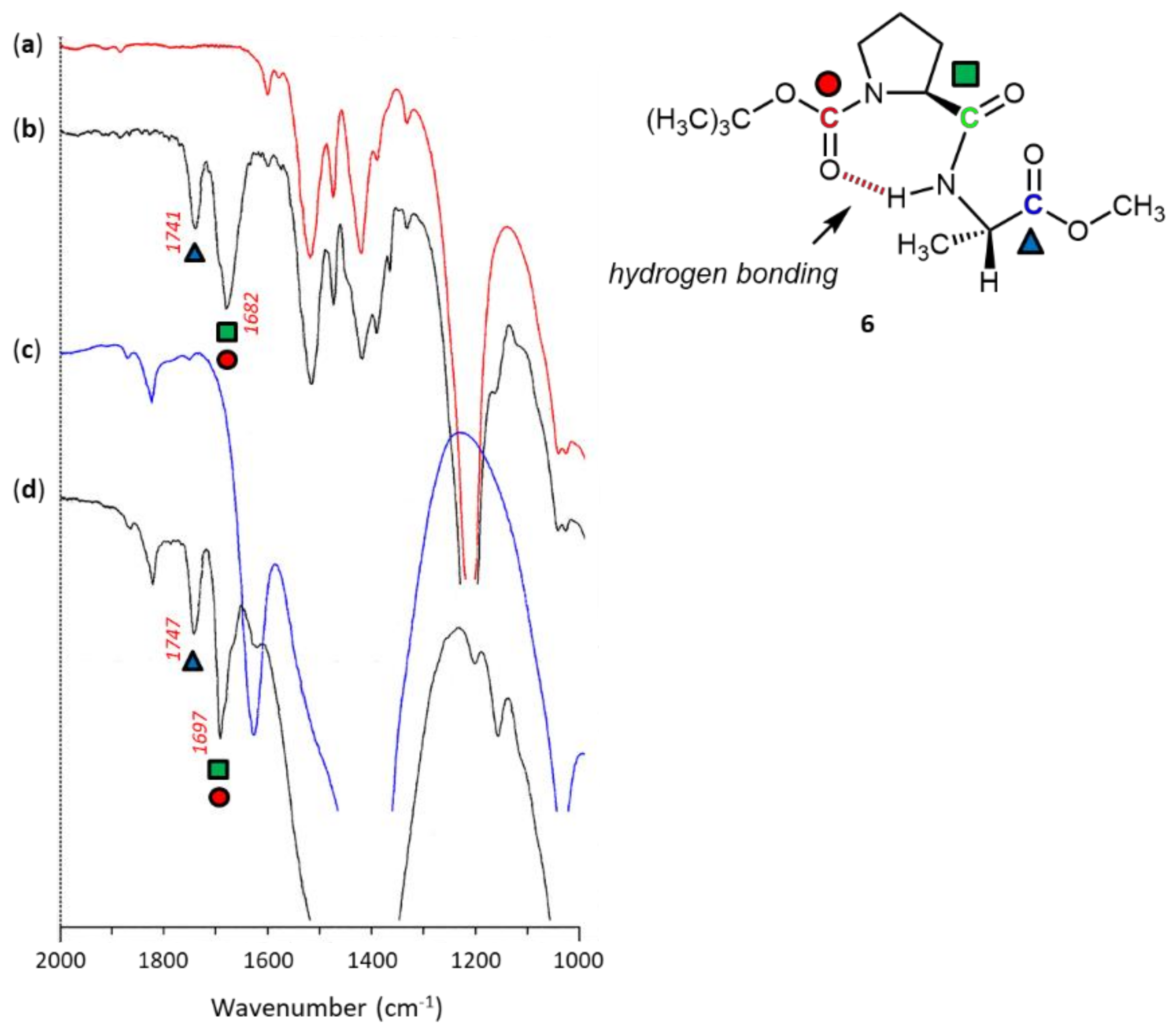
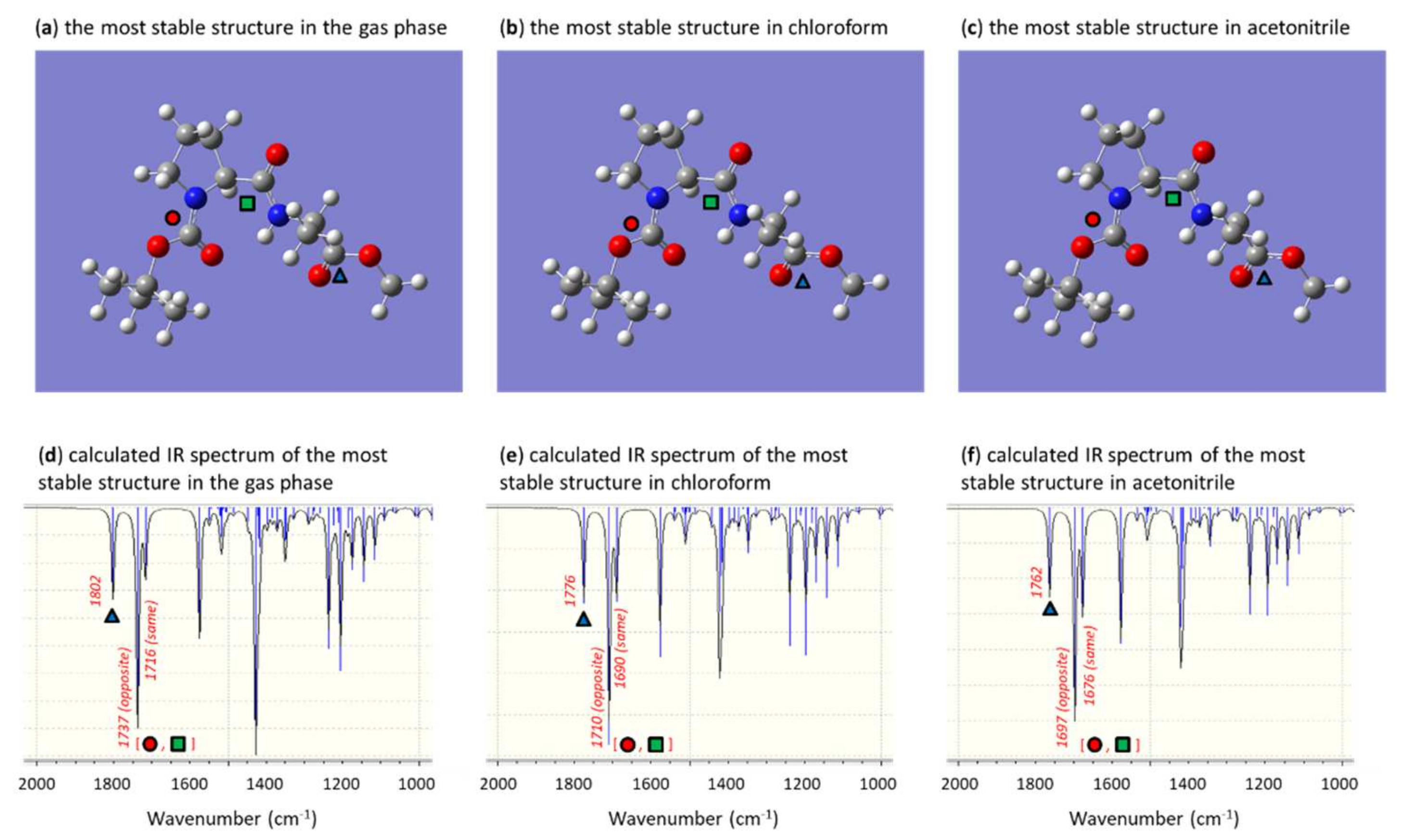
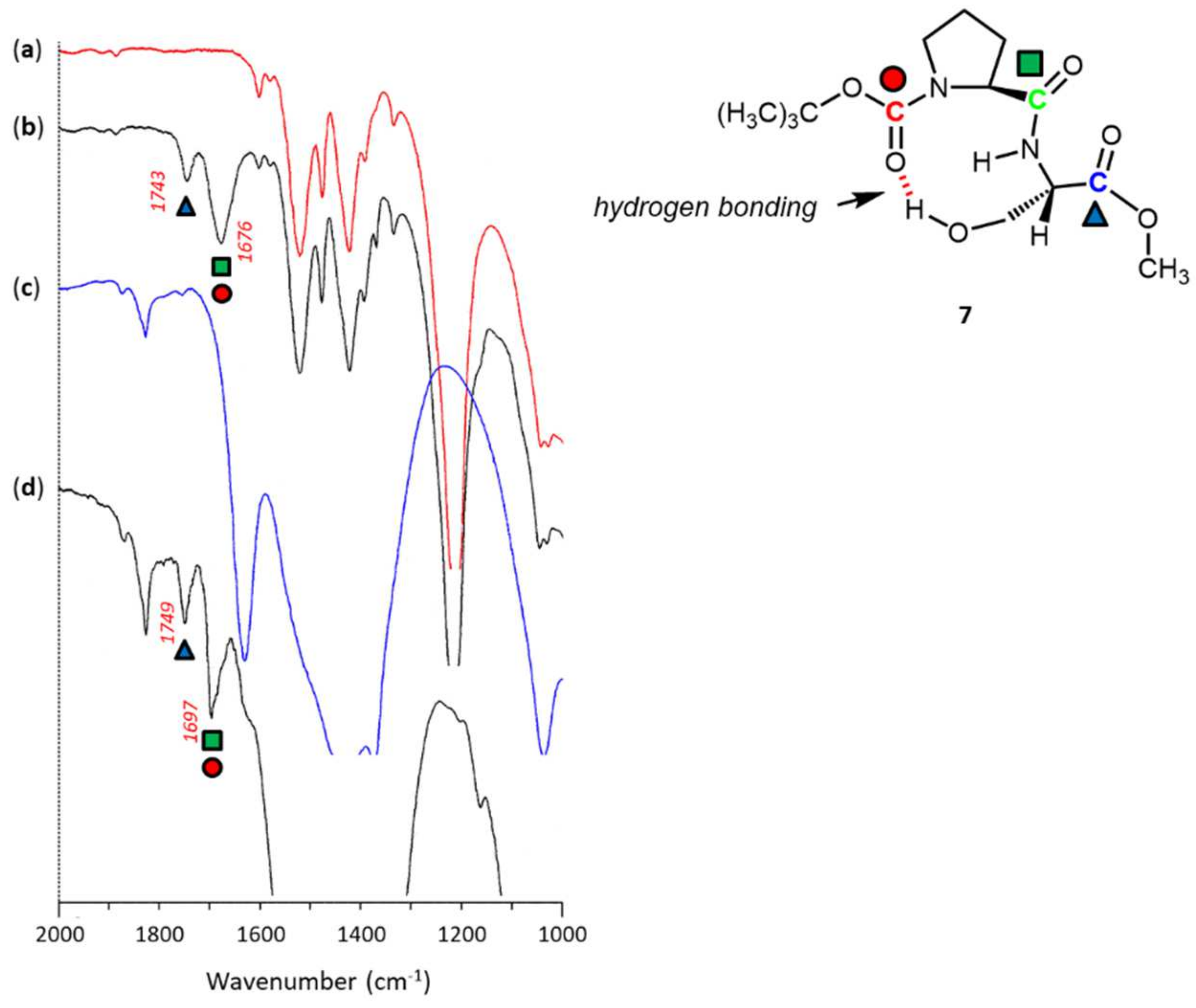
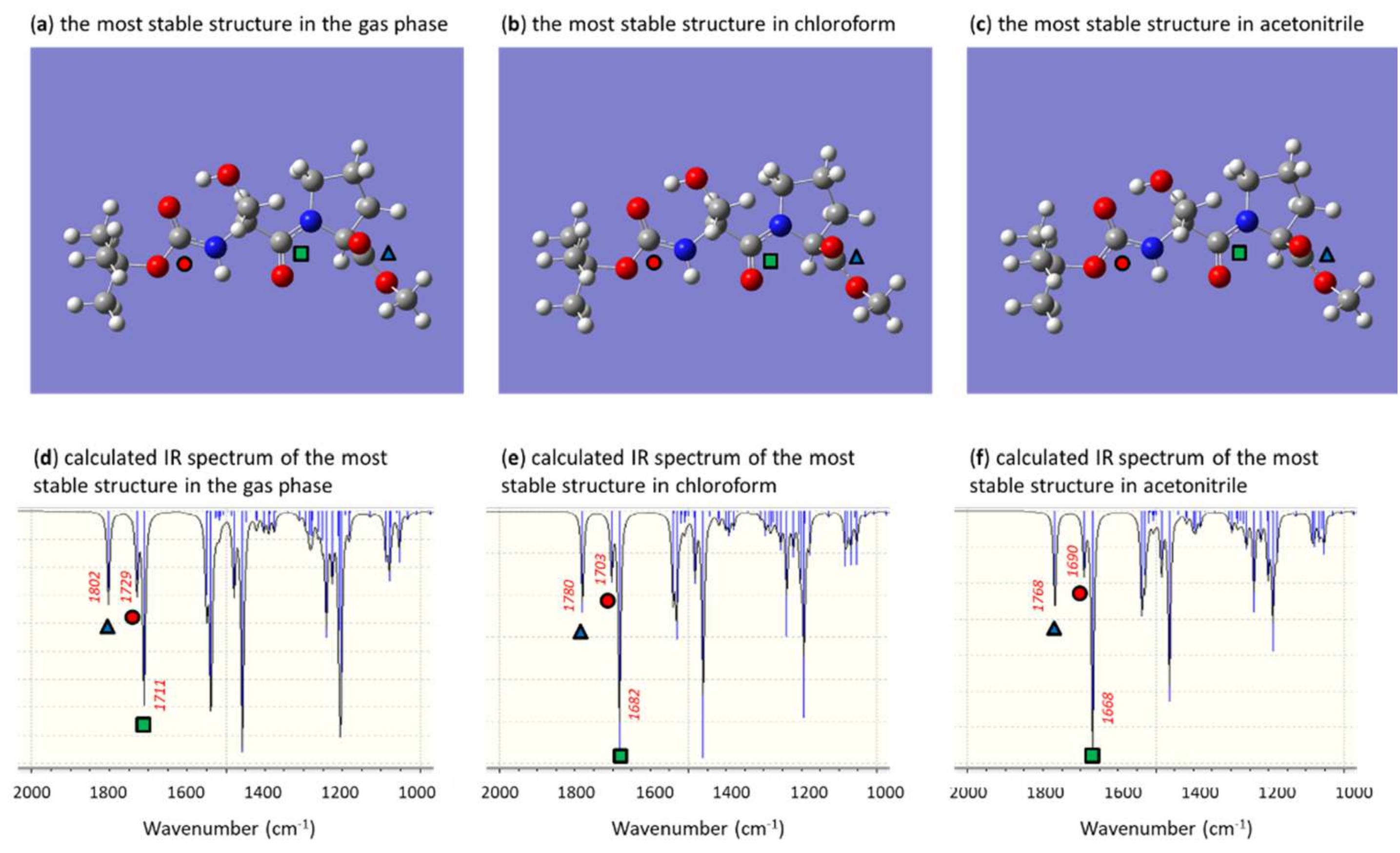
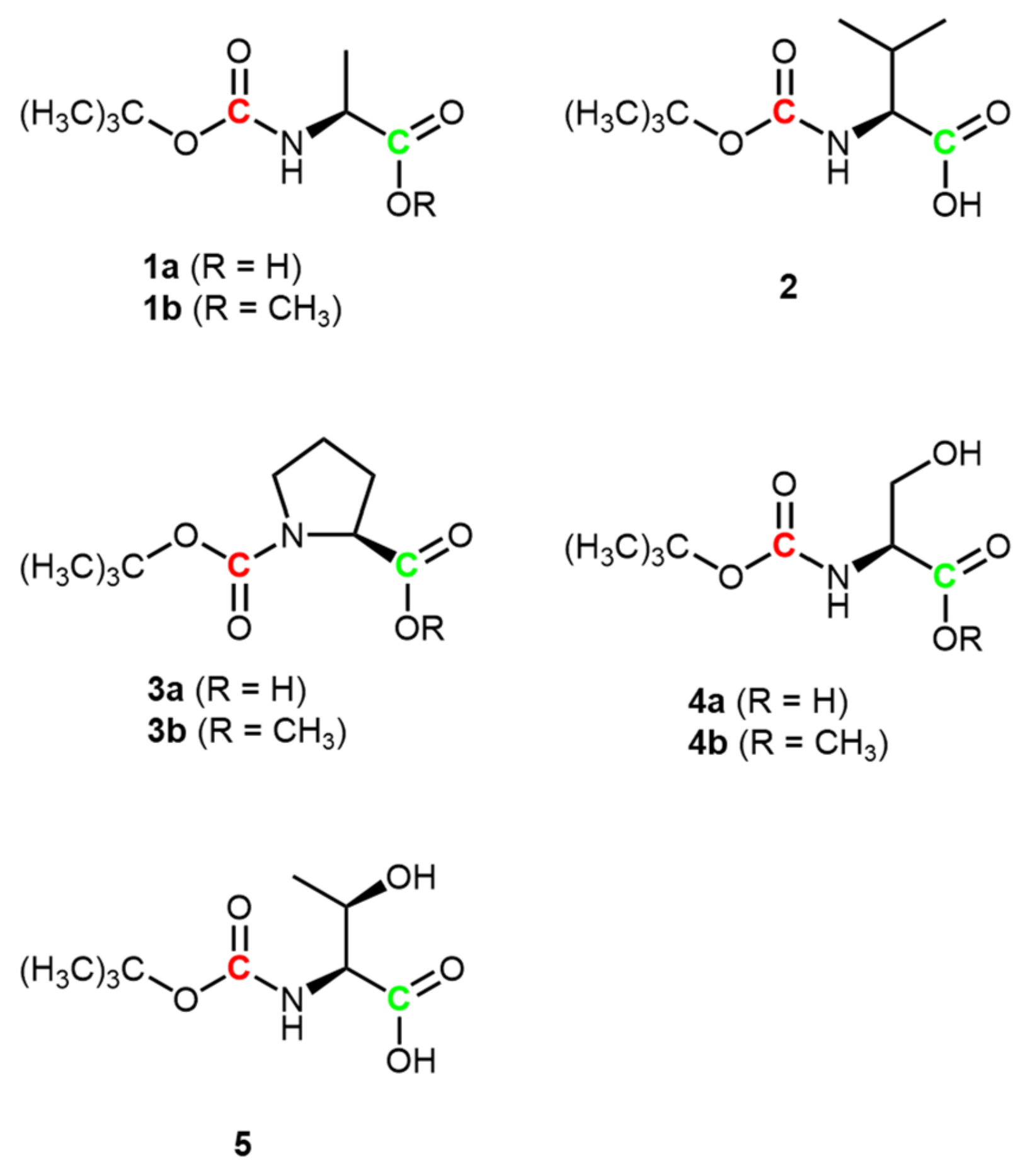
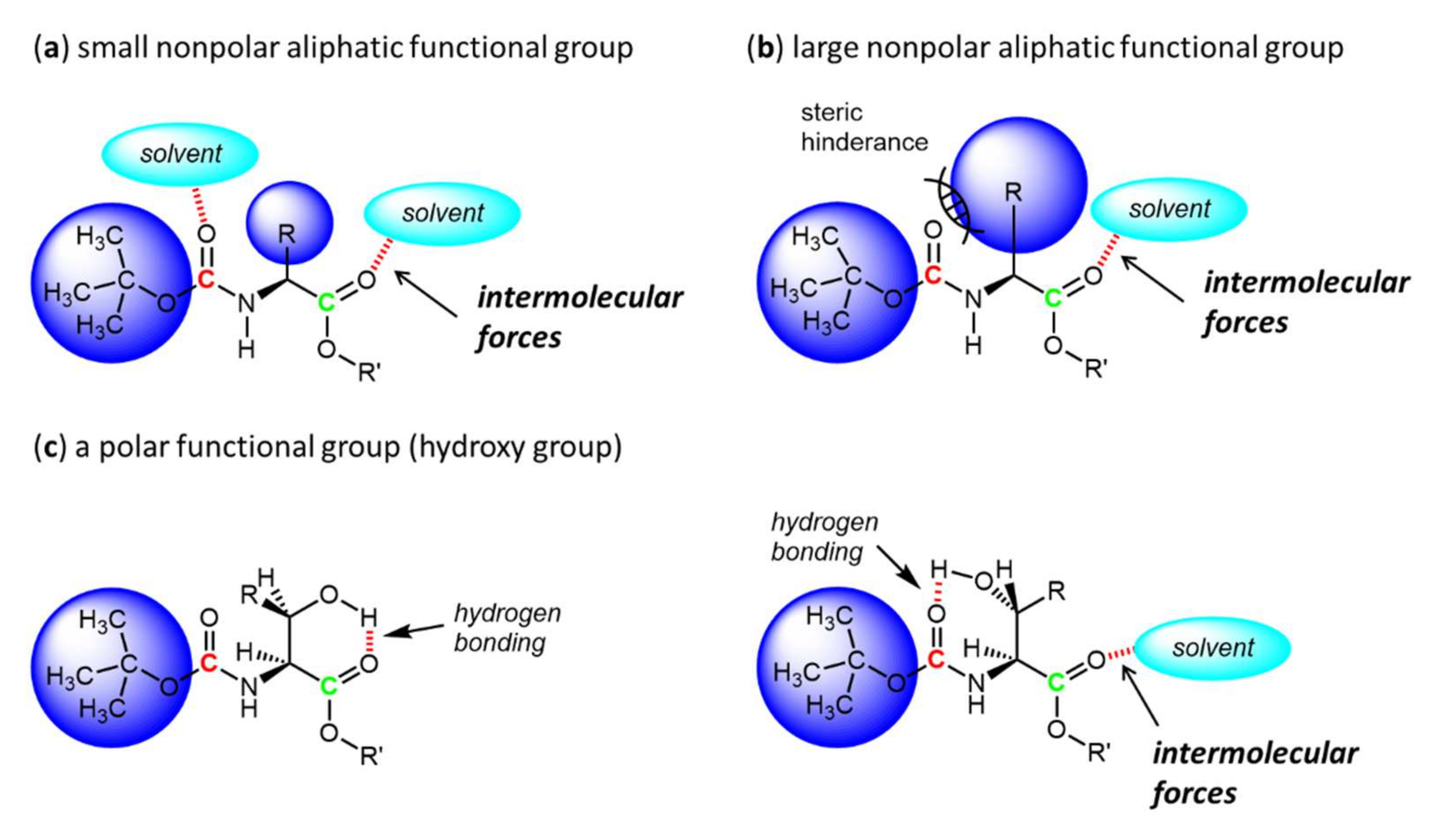
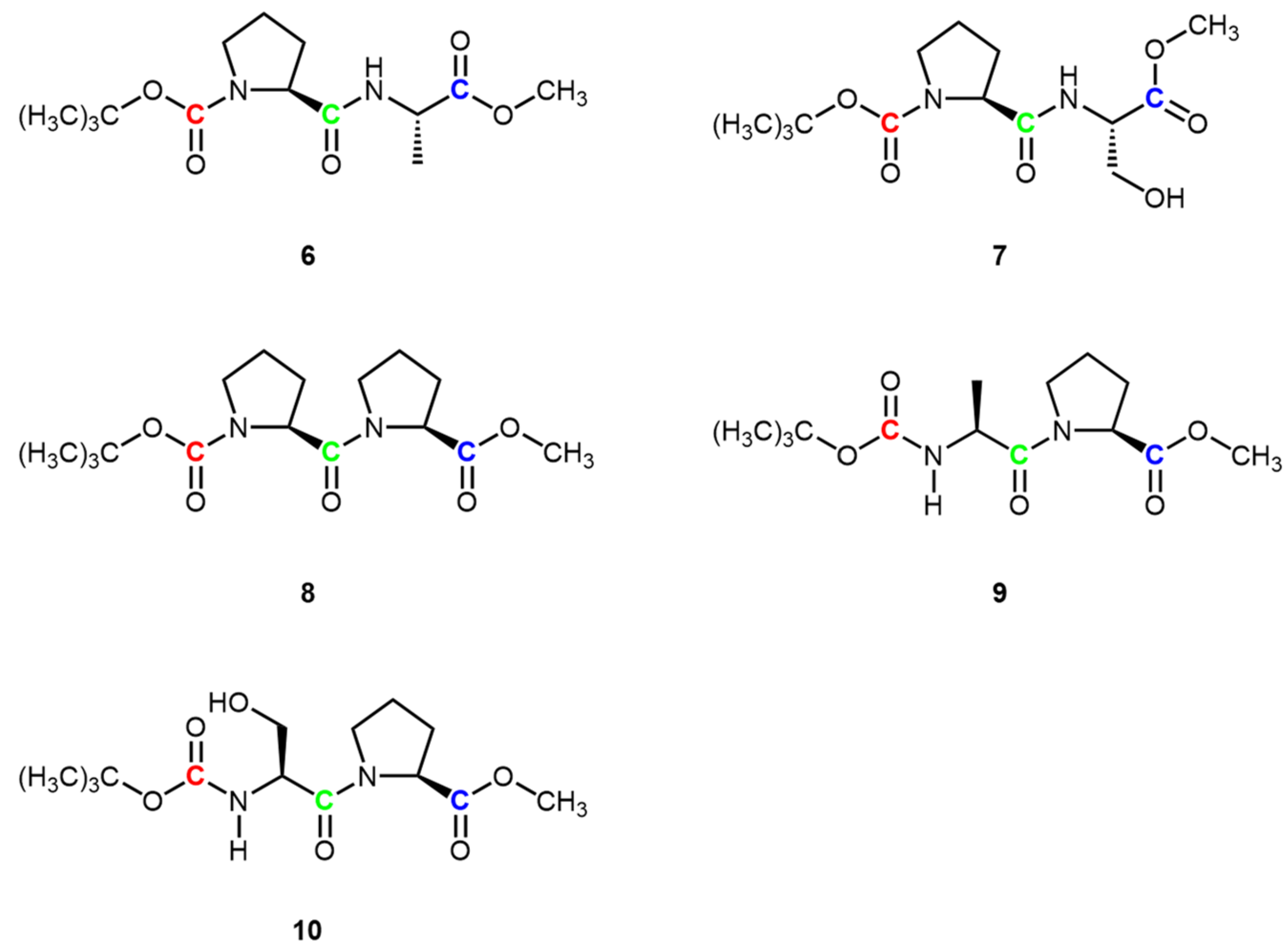
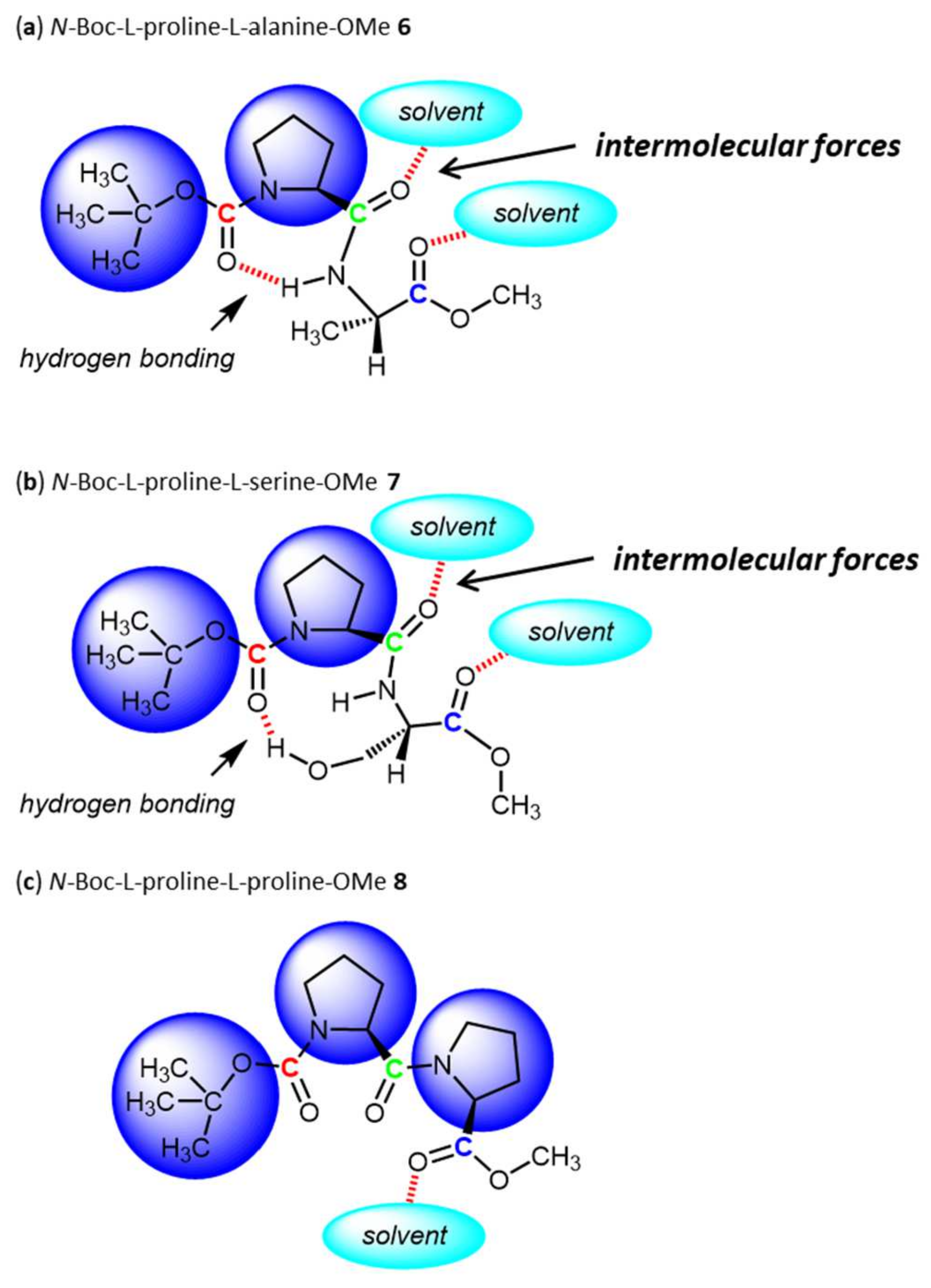
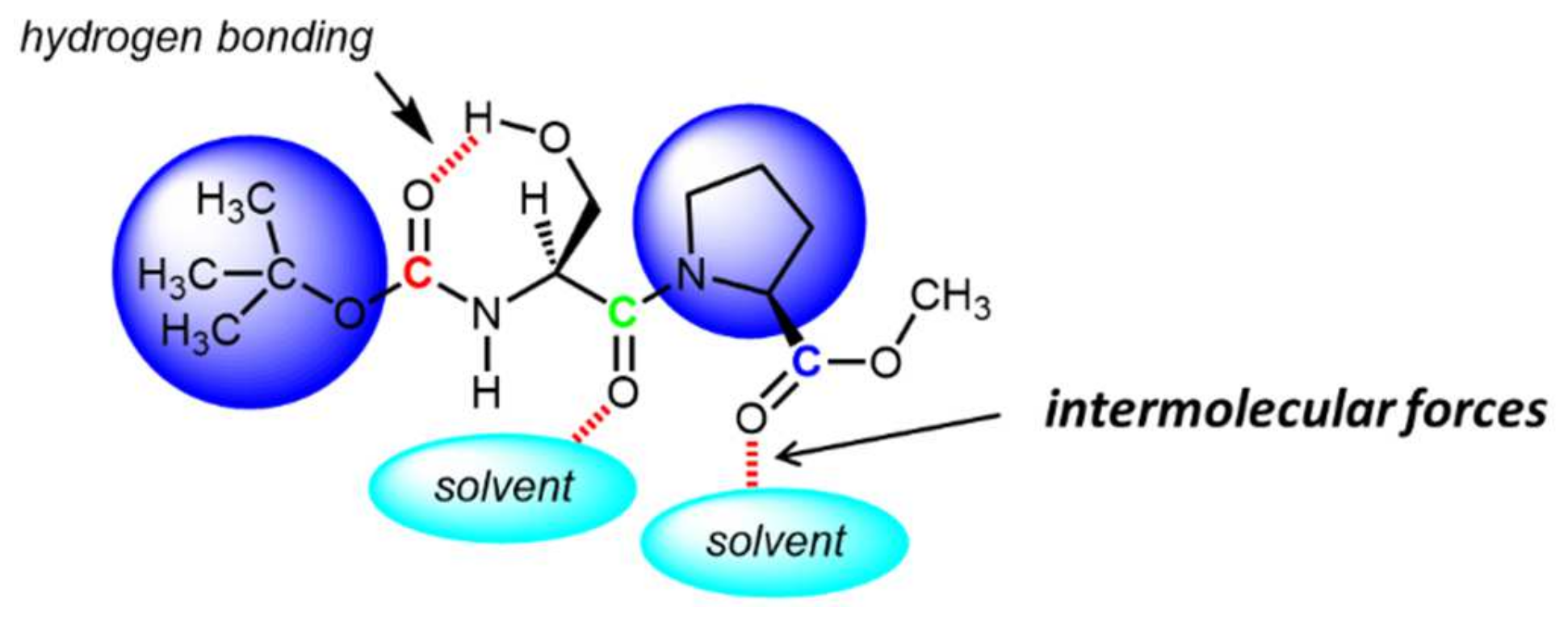
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwan, E.E.; Huang, S.G. Structural Elucidation with NMR Spectroscopy: Practical Strategies for Organic Chemists. Eur. J. Org. Chem. 2008, 2008, 2671–2688. [Google Scholar] [CrossRef]
- Günther, H. NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Dias, D.A.; Jones, O.A.H.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.-L.; Wishart, D.S. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef]
- Maeda, M.; Aoyama, T.; Takido, T.; Seno, M. Hydrogen-Bonding Interactions of C-Tetraundecenylresorc[4]arene with Some Guest Molecules. J. Oleo Sci. 2006, 55, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Mann, S.K.; Pham, T.N.; McQueen, L.L.; Lewandowski, J.R.; Brown, S.P. Revealing Intermolecular Hydrogen Bonding Structure and Dynamics in a Deep Eutectic Pharmaceutical by Magic-Angle Spinning NMR Spectroscopy. Mol. Pharm. 2020, 17, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milic, M.; Targos, K.; Chavez, M.T.; Thompson, M.A.M.; Jennings, J.J.; Franz, A.K. NMR Quantification of Hydrogen-Bond-Accepting Ability for Organic Molecules. J. Org. Chem. 2021, 86, 6031–6043. [Google Scholar] [CrossRef]
- Pease, L.G.; Deber, C.M.; Blout, E.R. Cyclic Peptides. V. 1H and 13C Nuclear Magnetic Resonance Determination of the Preferred β Conformation for Proline-Containing Cyclic Hexapeptides. J. Am. Chem. Soc. 1973, 95, 258–260. [Google Scholar] [CrossRef]
- Nakata, S.; Tenno, R.; Deguchi, A.; Yamamoto, H.; Hiraga, Y.; Izumi, S. Marangoni Flow around a Camphor Disk Regenerated by the Interaction between Camphor and Sodium Dodecyl Sulfate Molecules. Colloids Surf. A Physicochem. Eng. Asp. 2015, 466, 40–44. [Google Scholar] [CrossRef]
- Hiraga, Y.; Chaki, S.; Niwayama, S. 13C NMR Spectroscopic Studies of the Behaviors of Carbonyl Compounds in Various Solutions. Tetrahedron Lett. 2017, 58, 4677–4681. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Niwayama, S. Highly Efficient Selective Monohydrolysis of Symmetric Diesters. J. Org. Chem. 2000, 65, 5834–5836. [Google Scholar] [CrossRef]
- Sarnowski, M.P.; Kang, C.W.; Elbatrawi, Y.M.; Wojtas, L.; Valle, J.R.D. Peptide N-Amination Supports β-Sheet Conformation. Angew. Chem. Int. Ed. 2017, 56, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Ramotowska, S.; Zarzeczańska, D.; Dąbkowska, I.; Wcisło, A.; Niedziałkowski, P.; Czaczyk, E.; Grobelna, B.; Ossowski, T. Hydrogen Bonding and Protonation Effects in Amino Acids’ Anthraquinone Derivatives—Spectroscopic and Electrochemical Studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 222, 117226. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Interstrand Dipole-Dipole Interactions Can Stabilize the Collagen Triple Helix. J. Biol. Chem. 2011, 286, 22905–22912. [Google Scholar] [CrossRef] [Green Version]
- Shamsir, M.S.; Dalby, A.R. β-Sheet Containment by Flanking Prolines: Molecular Dynamic Simulations of the Inhibition of β-Sheet Elongation by Proline Residues in Human Prion Protein. Biophys. J. 2007, 92, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Kalmar, L.; Xue, B.; Tompa, P.; Daughdrill, G.W.; Uversky, V.N.; Han, K.-H. Contribution of Proline to the Pre-structuring Tendency of Transient Helical Secondary Structure Elements in Intrinsically Disordered Proteins. Biochim. Biophys. Acta 2014, 1840, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deber, C.M.; Bovey, F.A.; Carver, J.P.; Blout, E.R. Nuclear Magnetic Resonance Evidence for cis-Peptide Bonds in Proline Oligomers. J. Am. Chem. Soc. 1970, 92, 6191–6198. [Google Scholar] [CrossRef]
- Torchia, D.A.; Sparks, S.W. Proline Assignments and Identification of the Cis K116/P117 Peptide Bond in Liganded Staphylococcal Nuclease Using Isotope Edited 2D NMR Spectroscopy. J. Am. Chem. Soc. 1989, 111, 8315–8317. [Google Scholar] [CrossRef]
- Ohgo, K.; Dabalos, C.L.; Kumashiro, K.K. Solid-State NMR Spectroscopy and Isotopic Labeling Target Abundant Dipeptide Sequences in Elastin’s Hydrophobic Domains. Macromolecules 2018, 51, 2145–2156. [Google Scholar] [CrossRef]
- Lansing, J.C.; Hu, J.G.; Belenky, M.; Griffin, R.G.; Herzfeld, J. Solid-State NMR Investigation of the Buried X-Proline Peptide Bonds of Bacteriorhodopsin. Biochemistry 2003, 42, 3586–3593. [Google Scholar] [CrossRef]
- Aliv, A.E.; Courtier-Murias, D. Conformational Analysis of L-Prolines in Water. J. Phys. Chem. B 2007, 111, 14034–14042. [Google Scholar] [CrossRef]
- Budαěšínský, M.; Ragnarsson, U.; Lankiewicz, L.; Grehn, L.; Slaninová, J.; Hlaváček, J. Synthesis and Utilization of 13C and 15N Backbone-labeled Proline: NMR Study of Synthesized Oxytocin with Backbone-labeled C-terminal Tripeptide Amide. Amino Acids 2005, 29, 151–160. [Google Scholar] [CrossRef]
- Presser, A.; Hüfner, A. Trimethylsilyldiazomethane—A Mild and Efficient Reagent for the Methylation of Carboxylic Acids and Alcohols in Natural Products. Mon. Chem. 2004, 135, 1015–1022. [Google Scholar] [CrossRef]
- Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. Methylation of α-Amino Acids and Derivatives Using Trimethylsilyldiazomethane. Chem. Biol. Drug Des. 2009, 73, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Blatchly, R.A.; Allen, T.R.; Bergstrom, D.T.; Shinozaki, Y. Microscale Synthesis and Analysis of a Dipeptide. J. Chem. Educ. 1989, 66, 965–966. [Google Scholar] [CrossRef]
- Abdurragman, J.; Wahyuningrum, D.; Achmad, S.; Bundjali, B. Synthesis of Dipeptide Benzoylalanylglycine Methyl Ester and Corrosion Inhibitor Evaluation by Tafel Equation. Sains Malays. 2011, 40, 973–976. [Google Scholar]
- Available online: https://gaussian.com/glossary/g09/ (accessed on 30 December 2021).
- Eilme, A. Solvatochromic Probe in Molecular Solvents: Implicit Versus Explicit Solvent Model. Theor. Chem. Acc. 2014, 133, 1538. [Google Scholar] [CrossRef] [Green Version]
- Klamt, A.; Moya, C.; Palomar, J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. [Google Scholar] [CrossRef] [Green Version]


























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiraga, Y.; Chaki, S.; Uyama, Y.; Hoshide, R.; Karaki, T.; Nagata, D.; Yoshimoto, K.; Niwayama, S. 13C NMR Spectroscopic Studies of Intra- and Intermolecular Interactions of Amino Acid Derivatives and Peptide Derivatives in Solutions. Organics 2022, 3, 38-58. https://doi.org/10.3390/org3010003
Hiraga Y, Chaki S, Uyama Y, Hoshide R, Karaki T, Nagata D, Yoshimoto K, Niwayama S. 13C NMR Spectroscopic Studies of Intra- and Intermolecular Interactions of Amino Acid Derivatives and Peptide Derivatives in Solutions. Organics. 2022; 3(1):38-58. https://doi.org/10.3390/org3010003
Chicago/Turabian StyleHiraga, Yoshikazu, Saori Chaki, Yuri Uyama, Ryosuke Hoshide, Takumi Karaki, Daisuke Nagata, Kanji Yoshimoto, and Satomi Niwayama. 2022. "13C NMR Spectroscopic Studies of Intra- and Intermolecular Interactions of Amino Acid Derivatives and Peptide Derivatives in Solutions" Organics 3, no. 1: 38-58. https://doi.org/10.3390/org3010003