
Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research

 , and
, and
Abstract
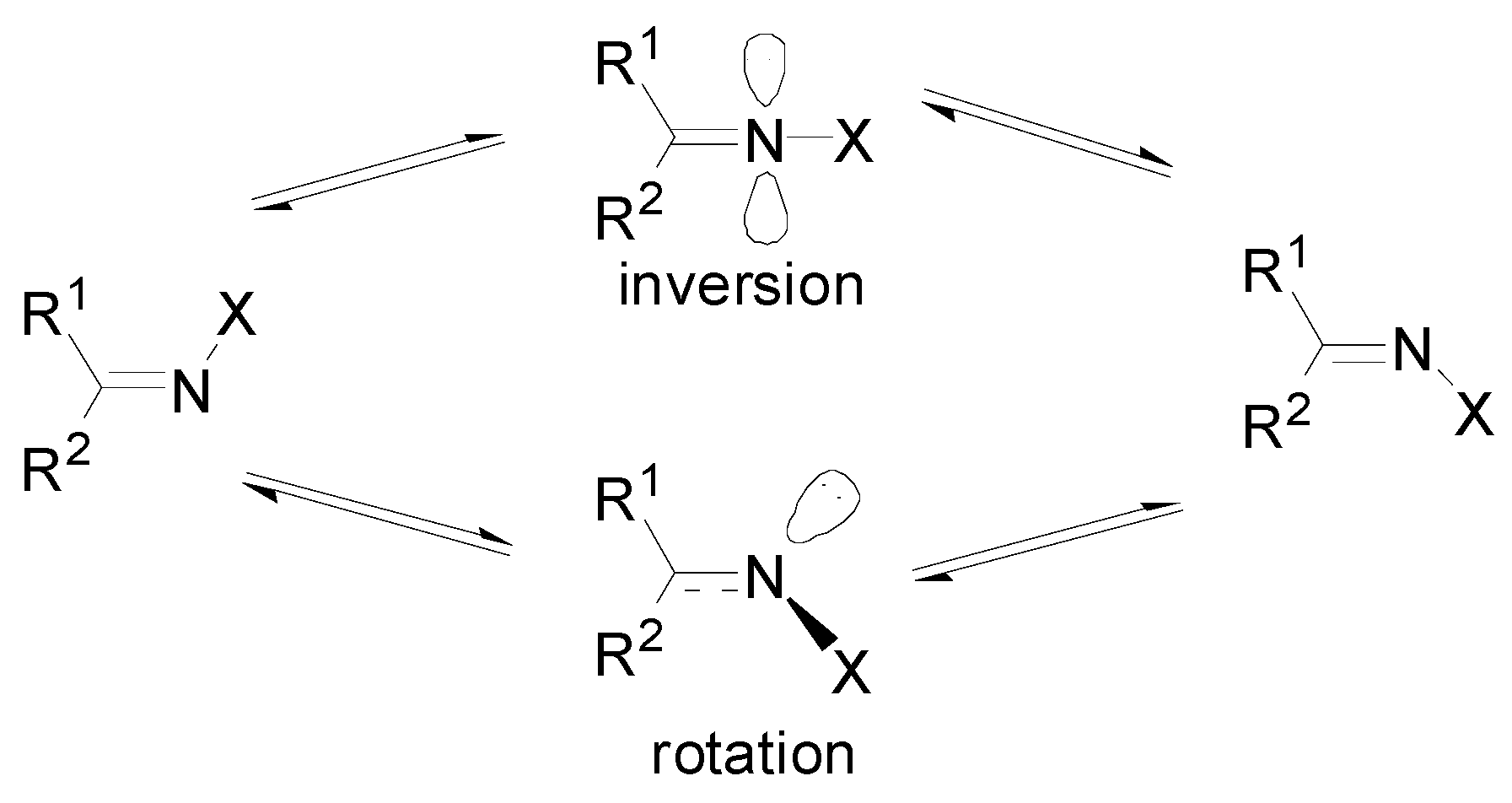
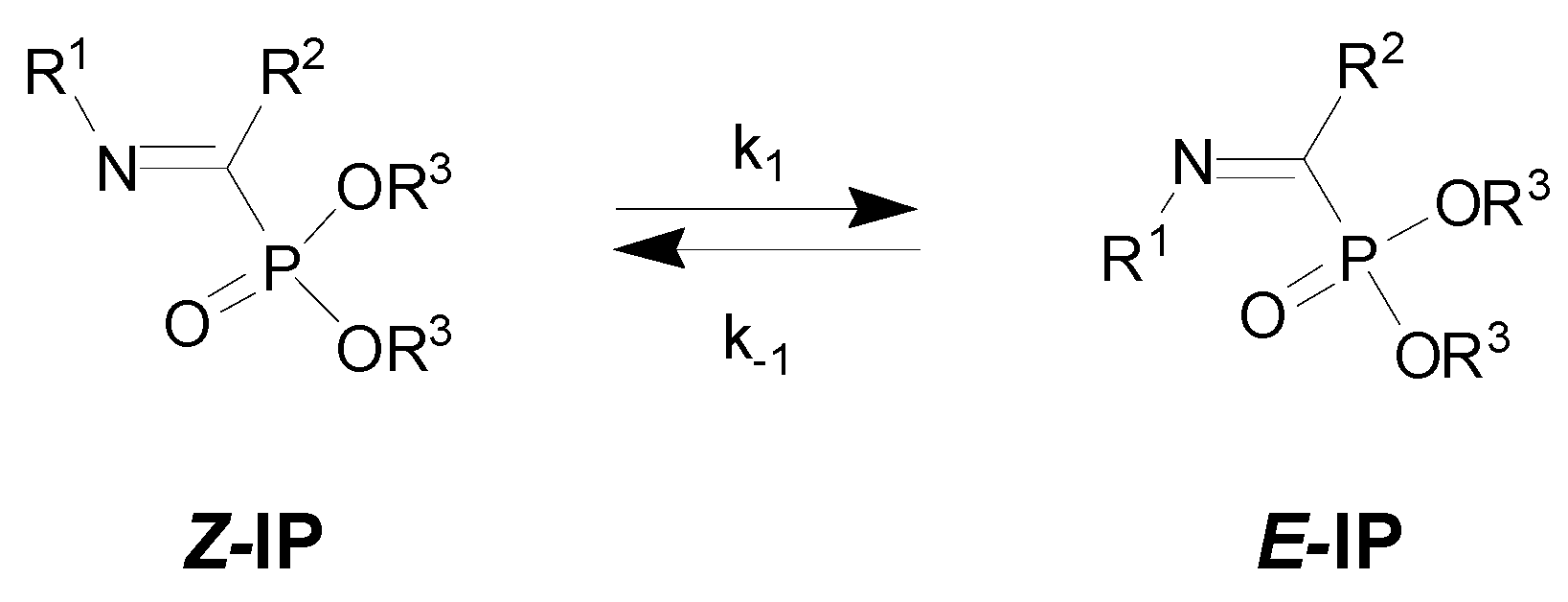
:1. Introduction
2. Methods of Calculations
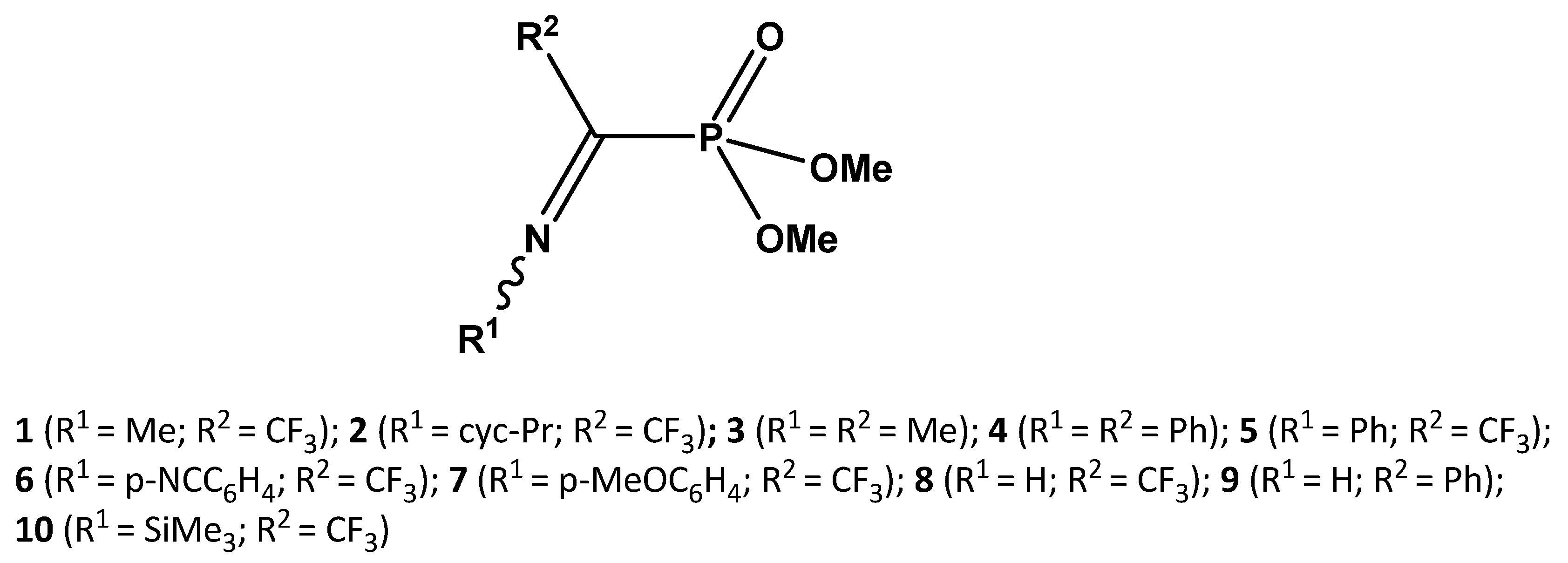
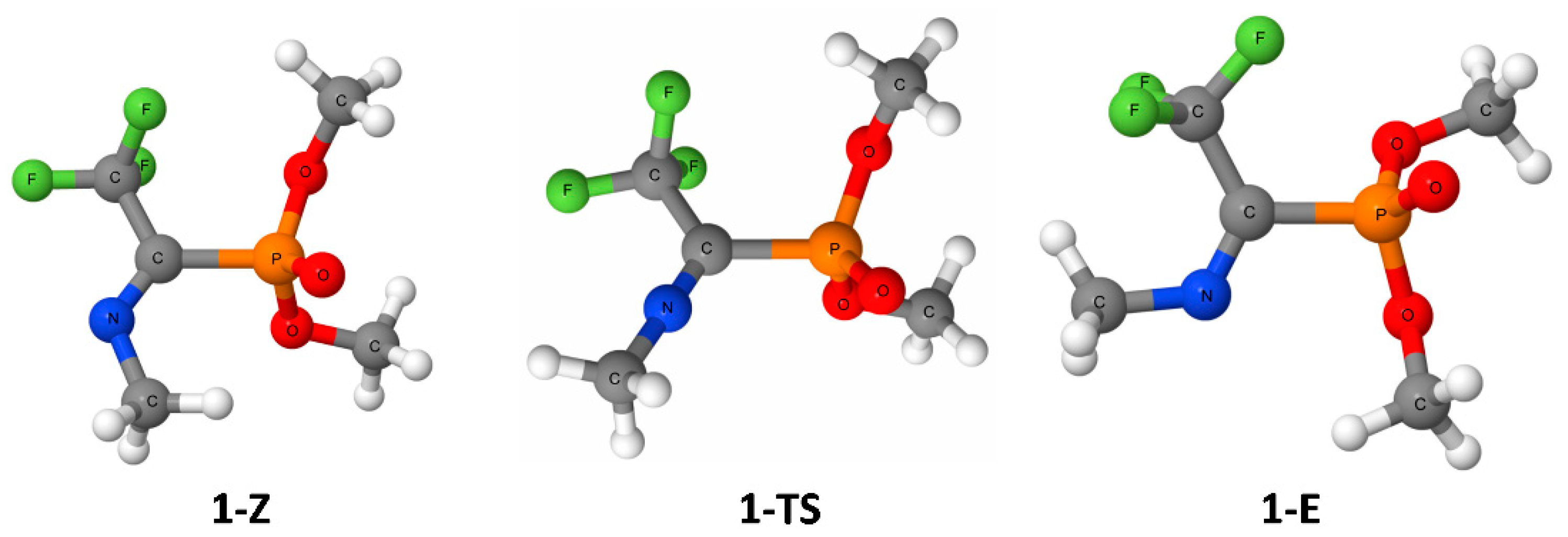
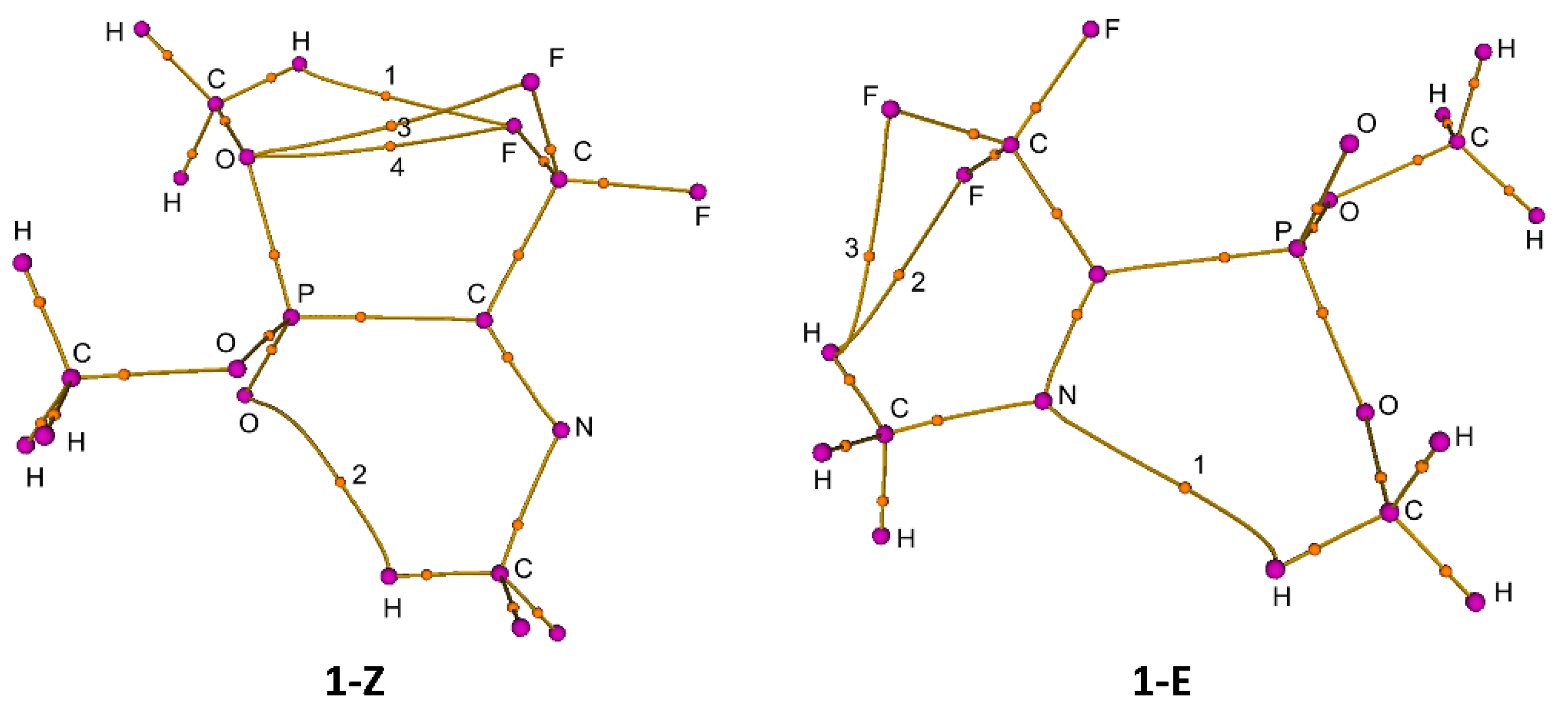
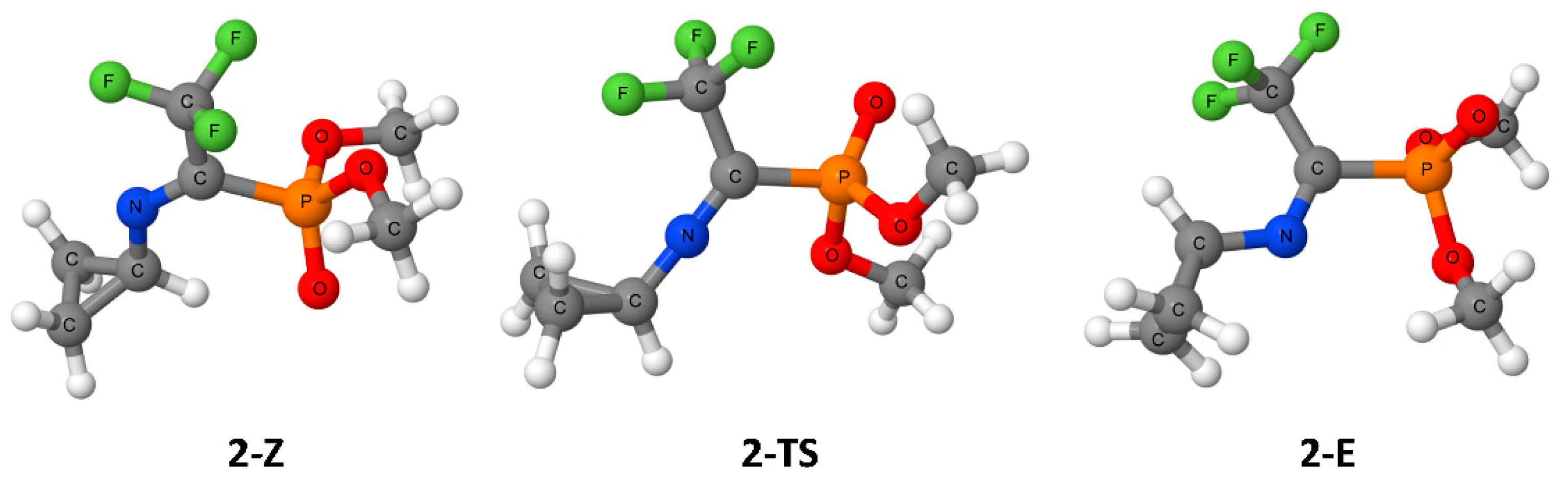
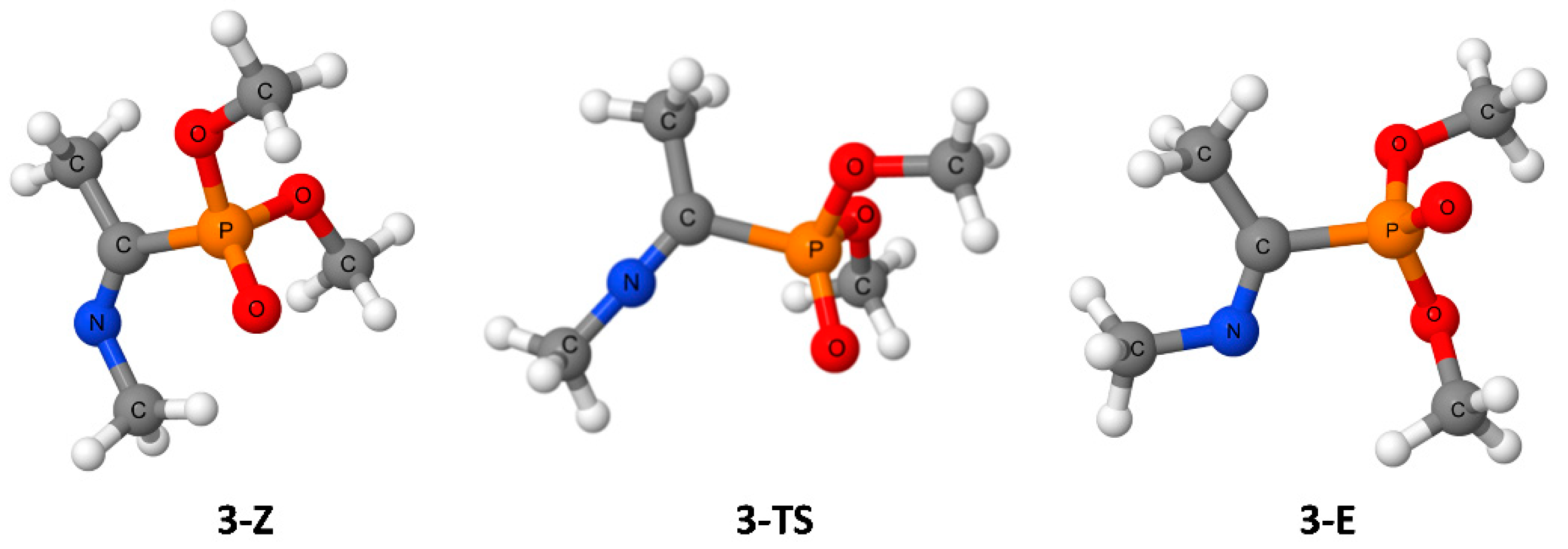
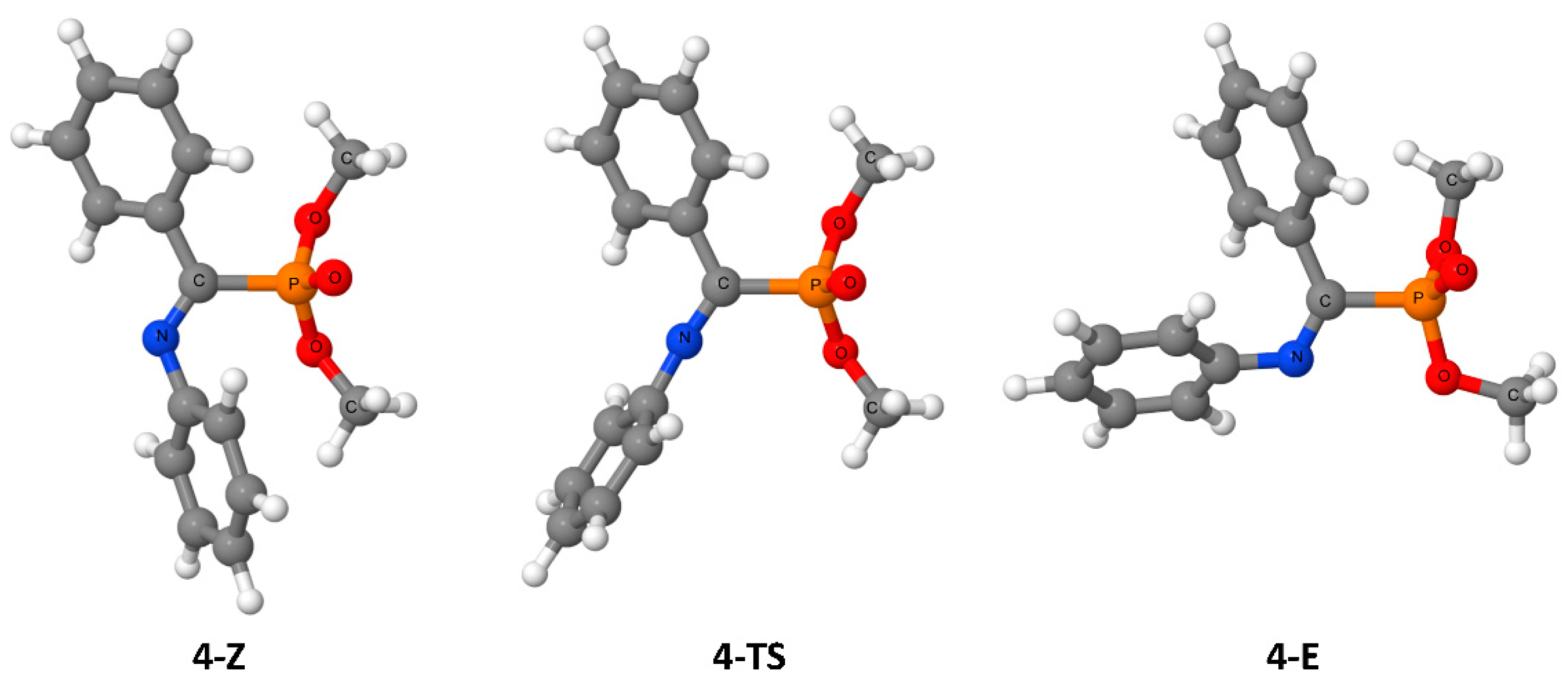
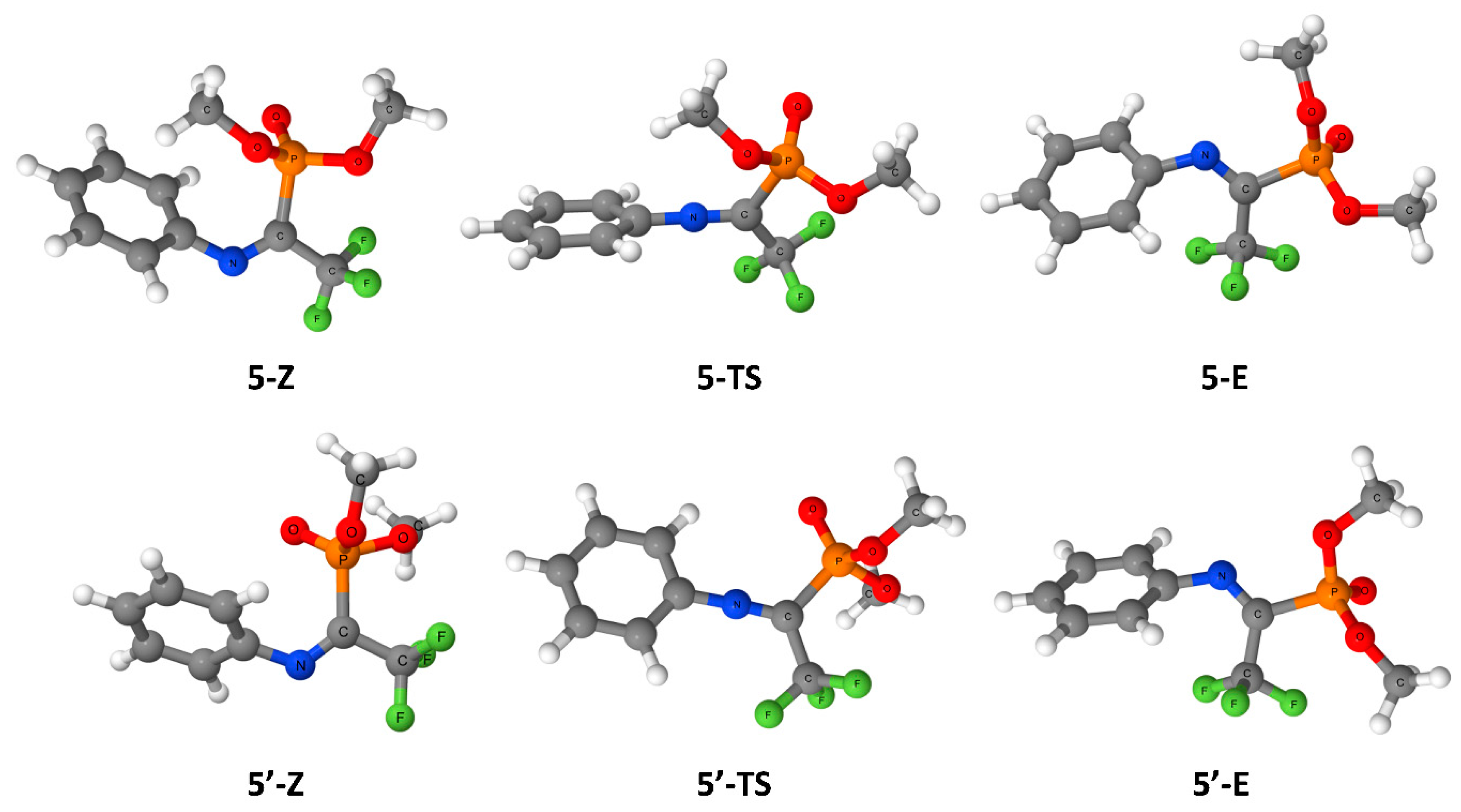
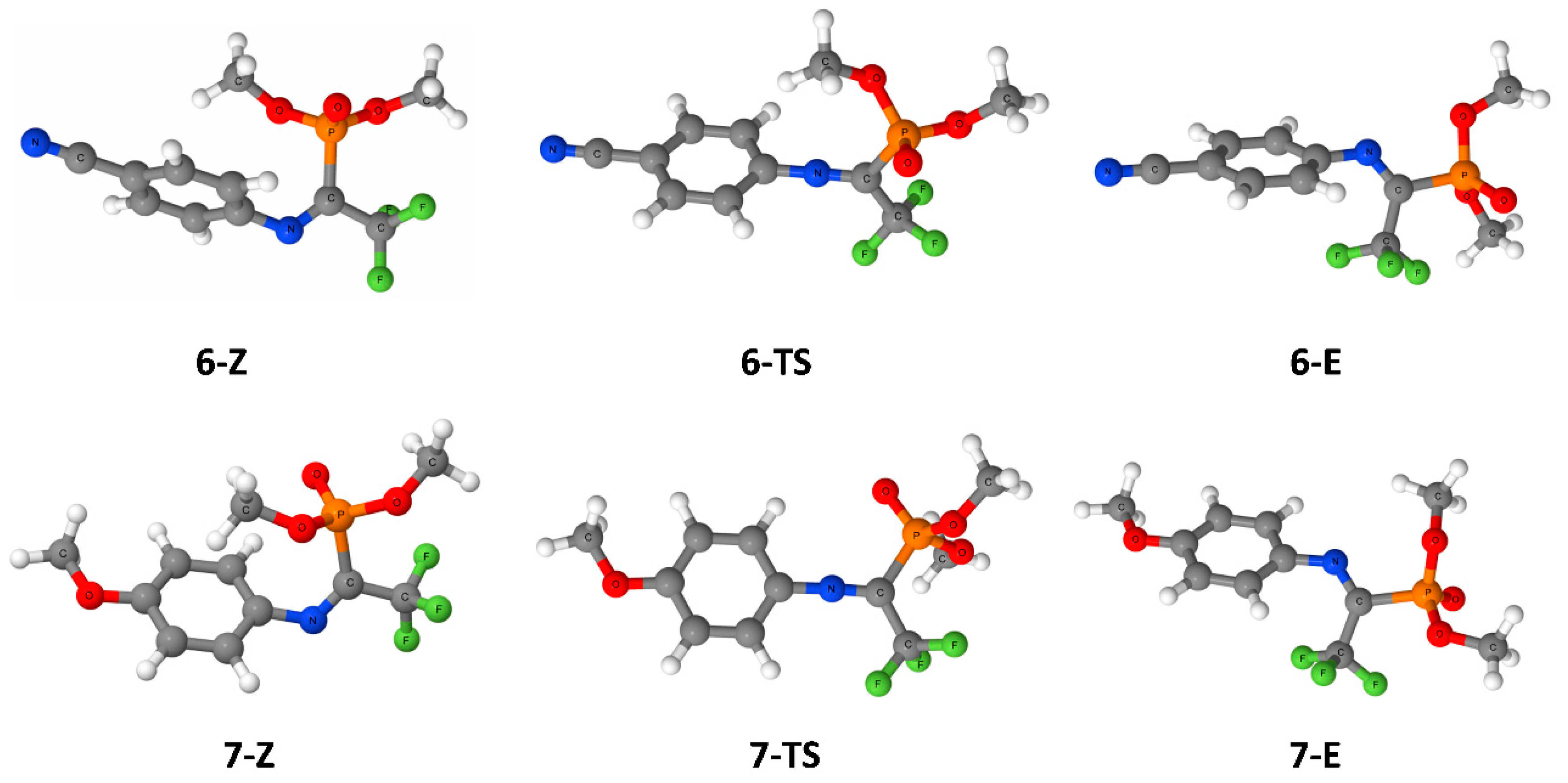
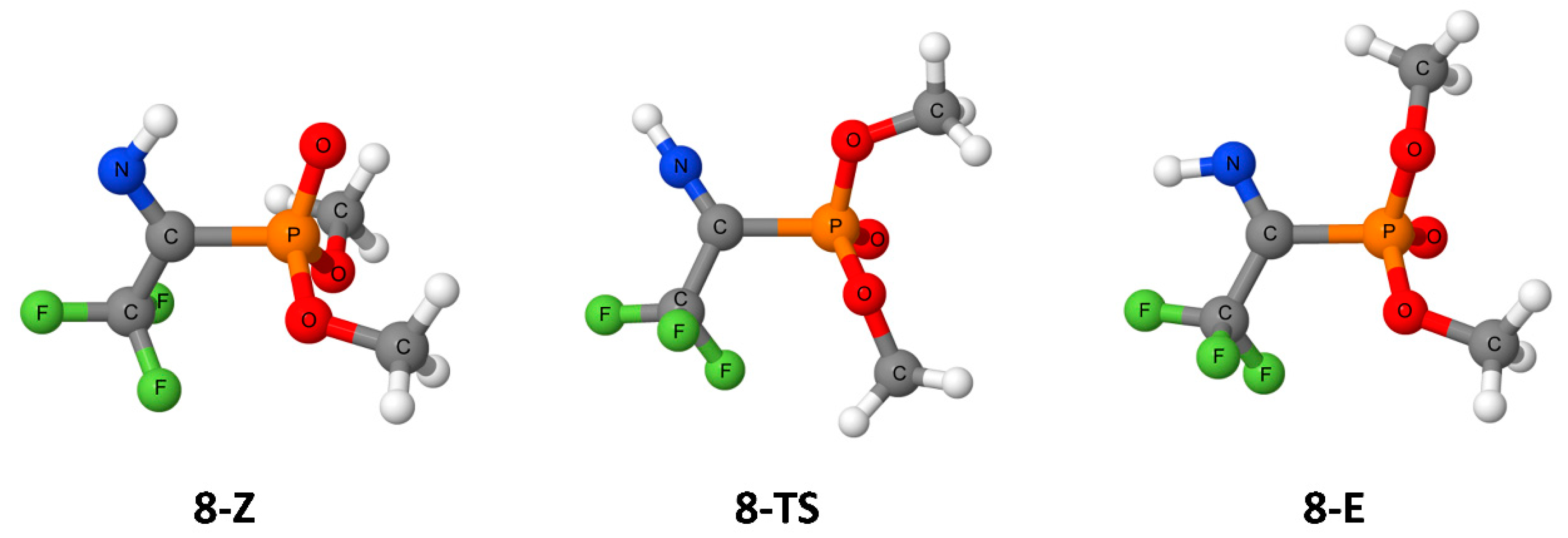
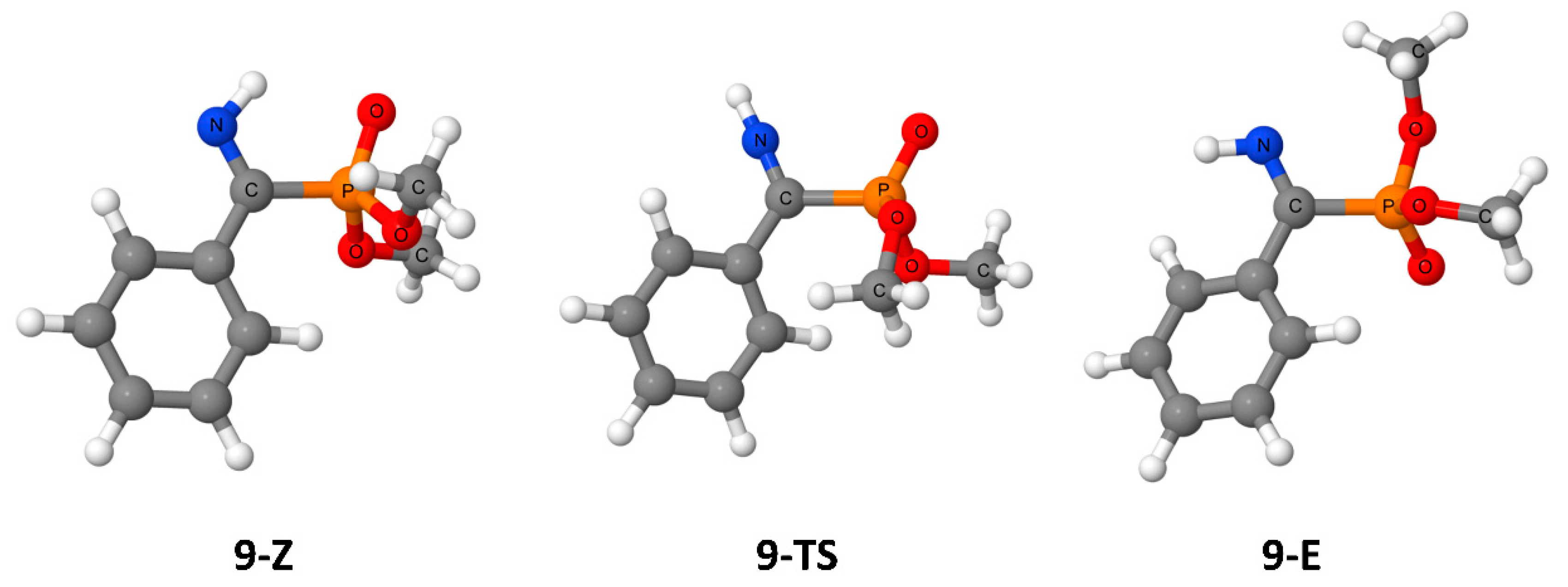
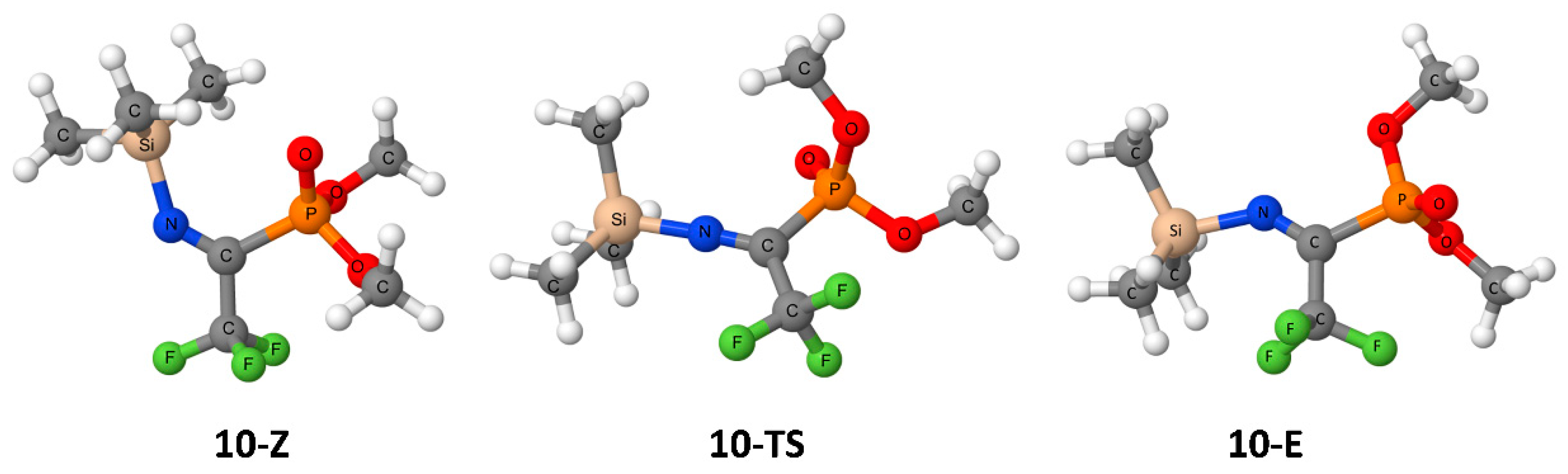
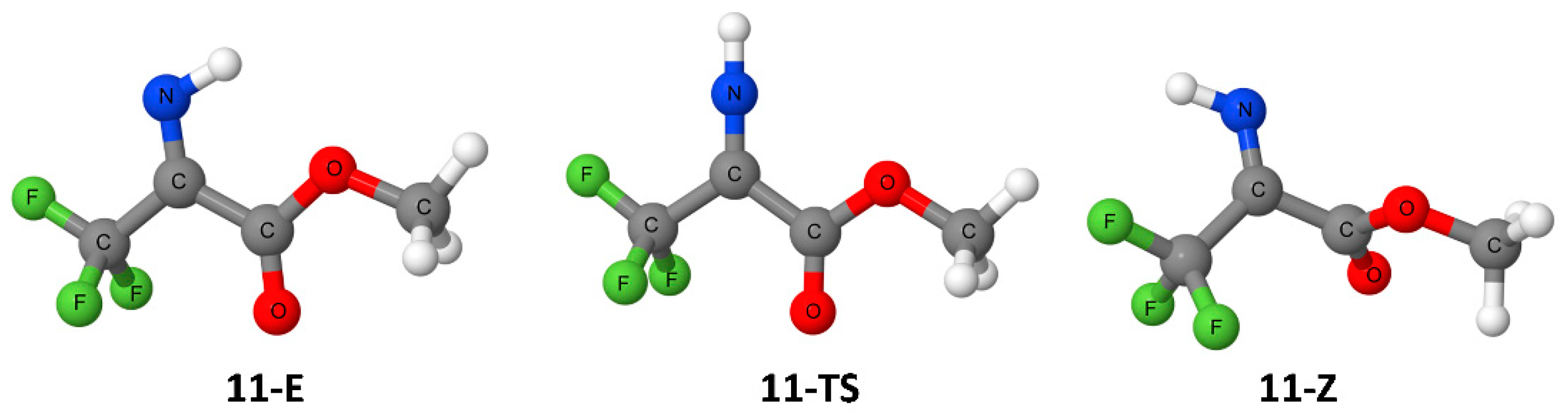
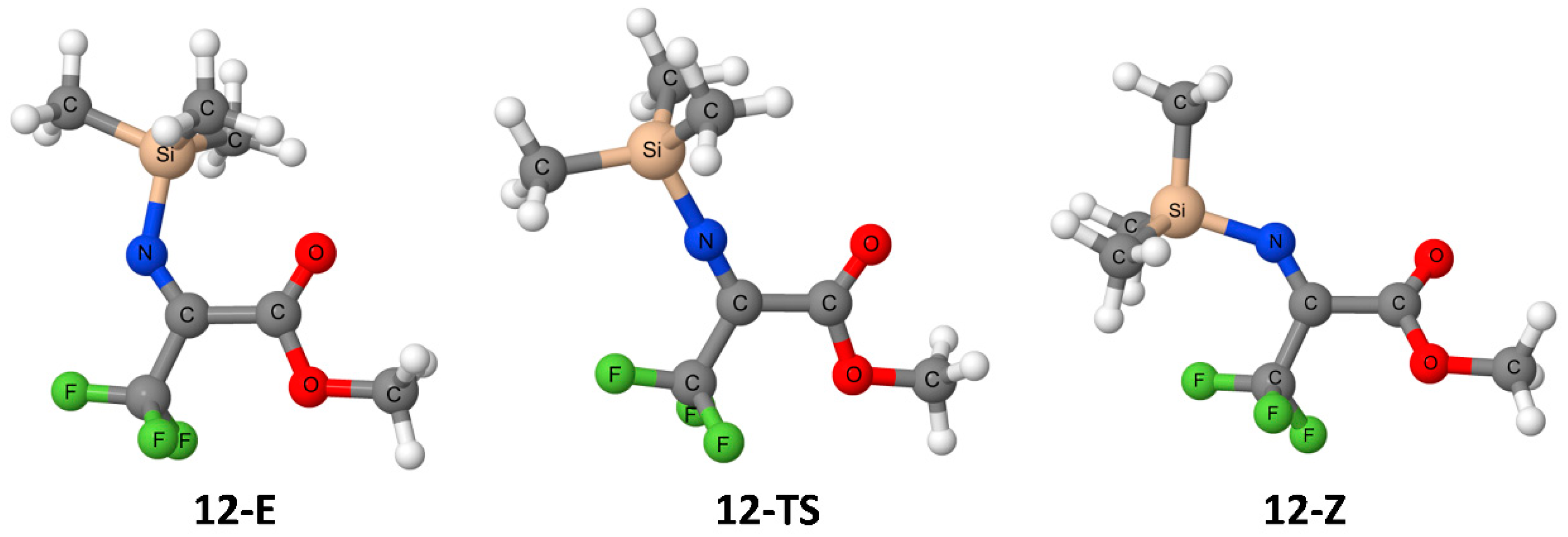
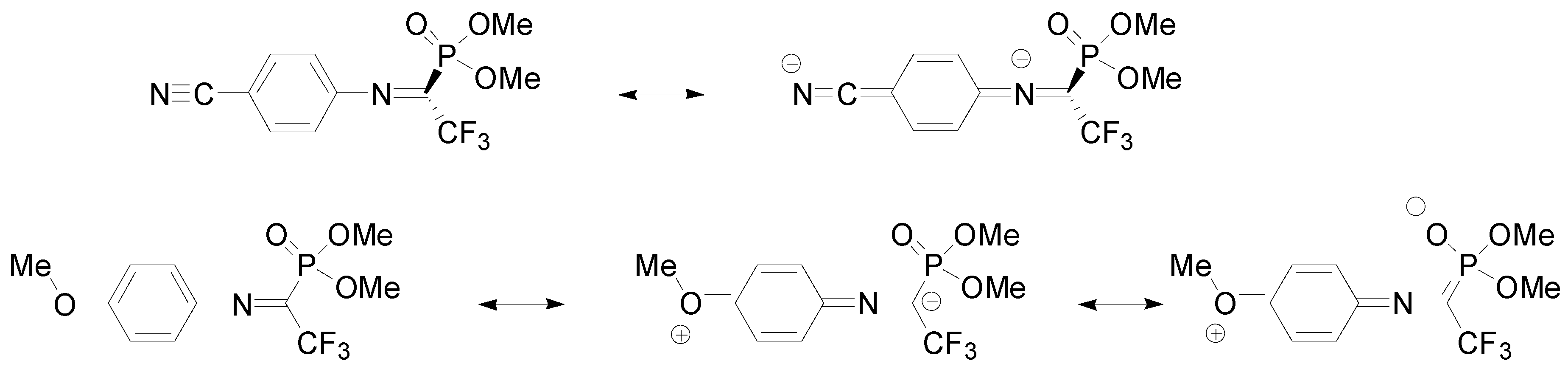
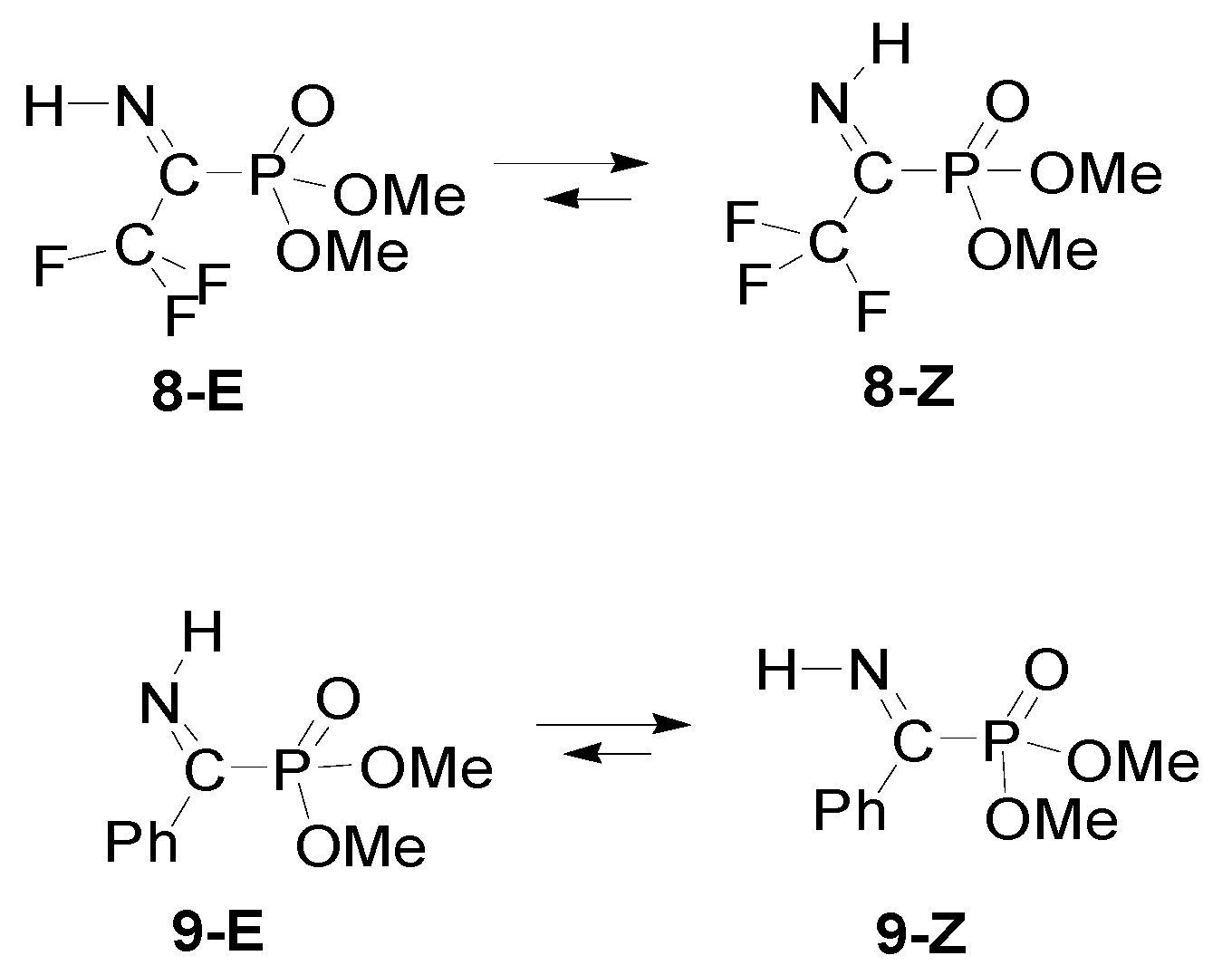
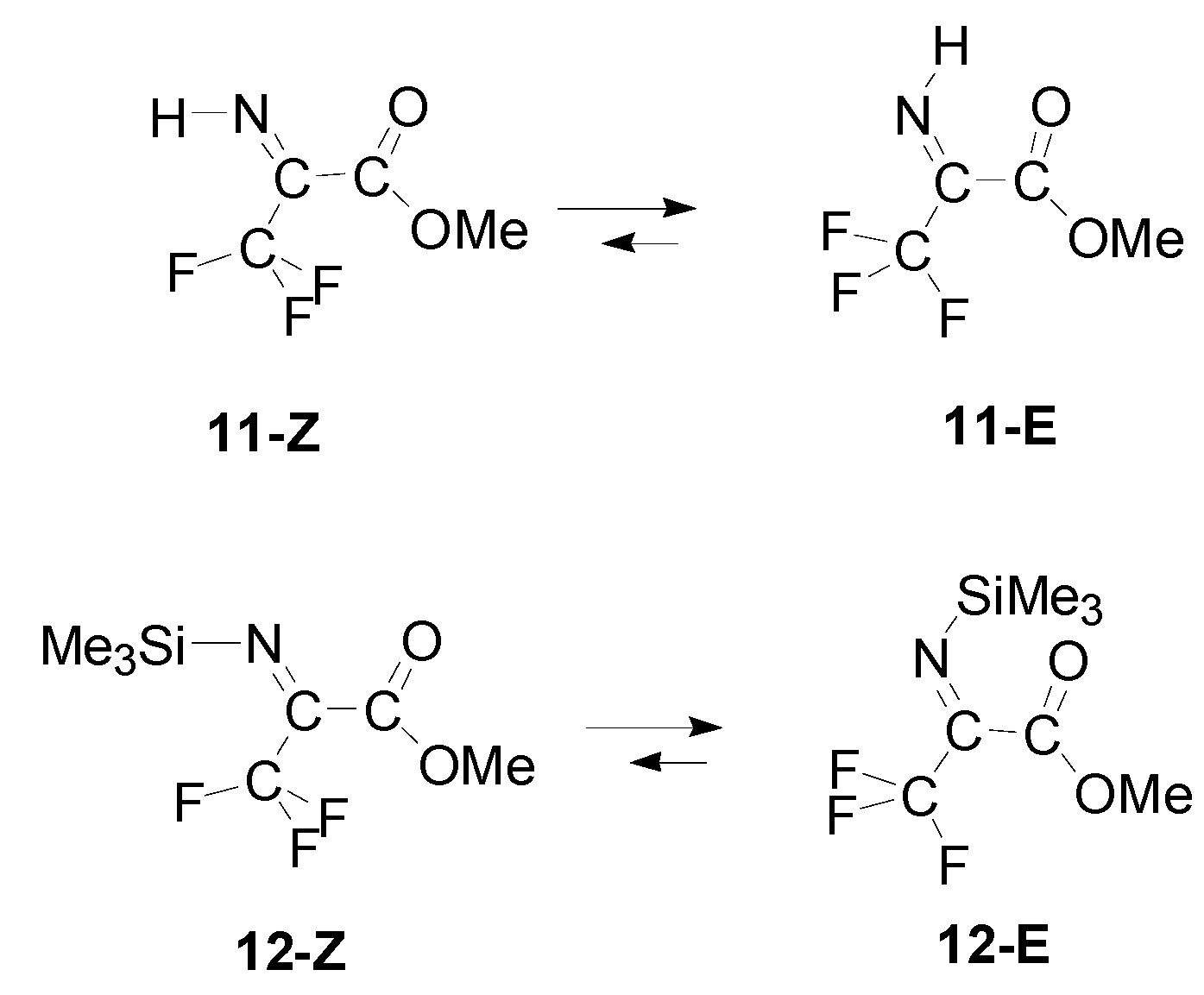
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kafarski, P.; Lejczak, B. Biological activity of aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Röschenthaler, G.-V.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Su, Q.; Li, Y.; Wang, B.; Liu, M.; Wang, H.; Wang, W.; Liu, F. Combining the Advantages of Alkene and Azo E–Z Photoisomerizations: Mechanistic Insights into Ketoimine Photoswitches. J. Phys. Chem. A 2017, 121, 2588–2596. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Knorr, R.; Ruhdorfer, J.; Mehlstäubl, J.; Böhrer, P.; Stephenson, D.S. (E, Z)1-Equilibria, 17 Demonstration of the Nitrogen Inversion Mechanism of Imines in a Schiff Base Model. Eur. J. Inorg. Chem. 1993, 126, 747–754. [Google Scholar] [CrossRef]
- Pirozhenko, V.V.; Rozhenko, A.B.; Avdeenko, A.P.; Konovalova, S.A.; Santalova, A.A. Z,E-Isomerization mechanism forN-arylthio-1,4-benzoquinonimines: DNMR and DFT investigations. Magn. Reson. Chem. 2008, 46, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J.; Guirado, A. A theoretical study of topomerization of imine systems: Inversion, rotation or mixed mechanisms? J. Comput. Chem. 2009, 31, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Khomutnik, Y.Y.; Onys’Ko, P.P.; Rassukanaya, Y.V.; Pirozhenko, V.V.; Sinitsa, A.D. N-aryltrifluoroacetimidoylphosphonates. Russ. J. Gen. Chem. 2013, 83, 445–452. [Google Scholar] [CrossRef]
- Roberts, J.D.; Hall, G.E.; Middleton, W.J. Nuclear magnetic resonance spectroscopy. Kinetics of isomerization of p-substituted hexafluoroacetone N-phenylimines. J. Am. Chem. Soc. 1971, 93, 4778–4781. [Google Scholar] [CrossRef]
- Onys’Ko, P.P.; Klyukovskii, D.V.; Bezdudnyi, A.V.; Pirozhenko, V.V.; Pustovit, Y.M.; Synytsya, A.D. N-(R-Cyclopropyl)trifluoroacetimidoyl Phosphonates. Phosphorus. Sulfur. Silicon Relat. Elem. 2014, 189, 1094–1101. [Google Scholar] [CrossRef]
- Onys’Ko, P.; Rassukana, Y.; Kolotylo, M.; Sinitsa, O.; Pirozhenko, V. α-Iminotrifluoroethylphosphonates: The First Representatives of N-H Imidoyl Phosphonates. Synthesis 2007, 2007, 2627–2630. [Google Scholar] [CrossRef]
- Wahbi, A.; Slimani, H.; Touil, S. Multinuclear NMR structural study of novel γ-iminophosphonate and phosphine oxide derivatives. J. Struct. Chem. 2015, 56, 34–41. [Google Scholar] [CrossRef]
- Ony‘skoP, P.; Chudakova, T.I.; Pirozhenko, V.V.; Rozhenko, A.B. α-Ketophosphonates in the Synthesis of α-iminophosphonates. Curr. Green Chem. 2020, 7, 226–238. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 21 April 2021).
- Canepa, P.; Hanson, R.M.; Ugliengo, P.; Alfredsson, M. J-ICE: A newJmolinterface for handling and visualizing crystallographic and electronic properties. J. Appl. Crystallogr. 2010, 44, 225–229. [Google Scholar] [CrossRef]
- TURBOMOLE GmbH. TURBOMOLE V6.3, 2011, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 21 April 2021).
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Haase, F.; Ahlrichs, R. Semidirect MP2 gradient evaluation on workstation computers: The MPGRAD program. J. Comput. Chem. 1993, 14, 907–912. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Van Maaren, P.J.; Ditz, J.C.; Lindh, R.; Van Der Spoel, D. Large-scale calculations of gas phase thermochemistry: Enthalpy of formation, standard entropy, and heat capacity. J. Chem. Phys. 2016, 145, 114305. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Hättig, C.; Schmitz, G.; Kossmann, J. Auxiliary basis sets for density-fitted correlated wavefunction calculations: Weighted core-valence and ECP basis sets for post-d elements. Phys. Chem. Chem. Phys. 2012, 14, 6549–6555. [Google Scholar] [CrossRef] [PubMed]
- Bushmarinov, I.; Lyssenko, K.A.; Antipin, M.Y. Atomic energy in the ’Atoms in Molecules’ theory and its use for solving chemical problems. Russ. Chem. Rev. 2009, 78, 283–302. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Hanson, R.M. Jmol– a paradigm shift in crystallographic visualization. J. Appl. Crystallogr. 2010, 43, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Ramasubbu, N.; Parthasarathy, R.; Murray-Rust, P. Angular preferences of intermolecular forces around halogen centers: Preferred directions of approach of electrophiles and nucleophiles around carbon-halogen bond. J. Am. Chem. Soc. 1986, 108, 4308–4314. [Google Scholar] [CrossRef]
- Lommerse, J.P.M.; Stone, A.J.; Taylor, R.; Allen, F.H. The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen. J. Am. Chem. Soc. 1996, 118, 3108–3116. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. The fluorine atom as a halogen bond donor, viz. a positive site. CrystEngComm 2011, 13, 6593–6596. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazian, S. Why Bond Critical Points Are Not “Bond” Critical Points. Chem. A Eur. J. 2018, 24, 5401–5405. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
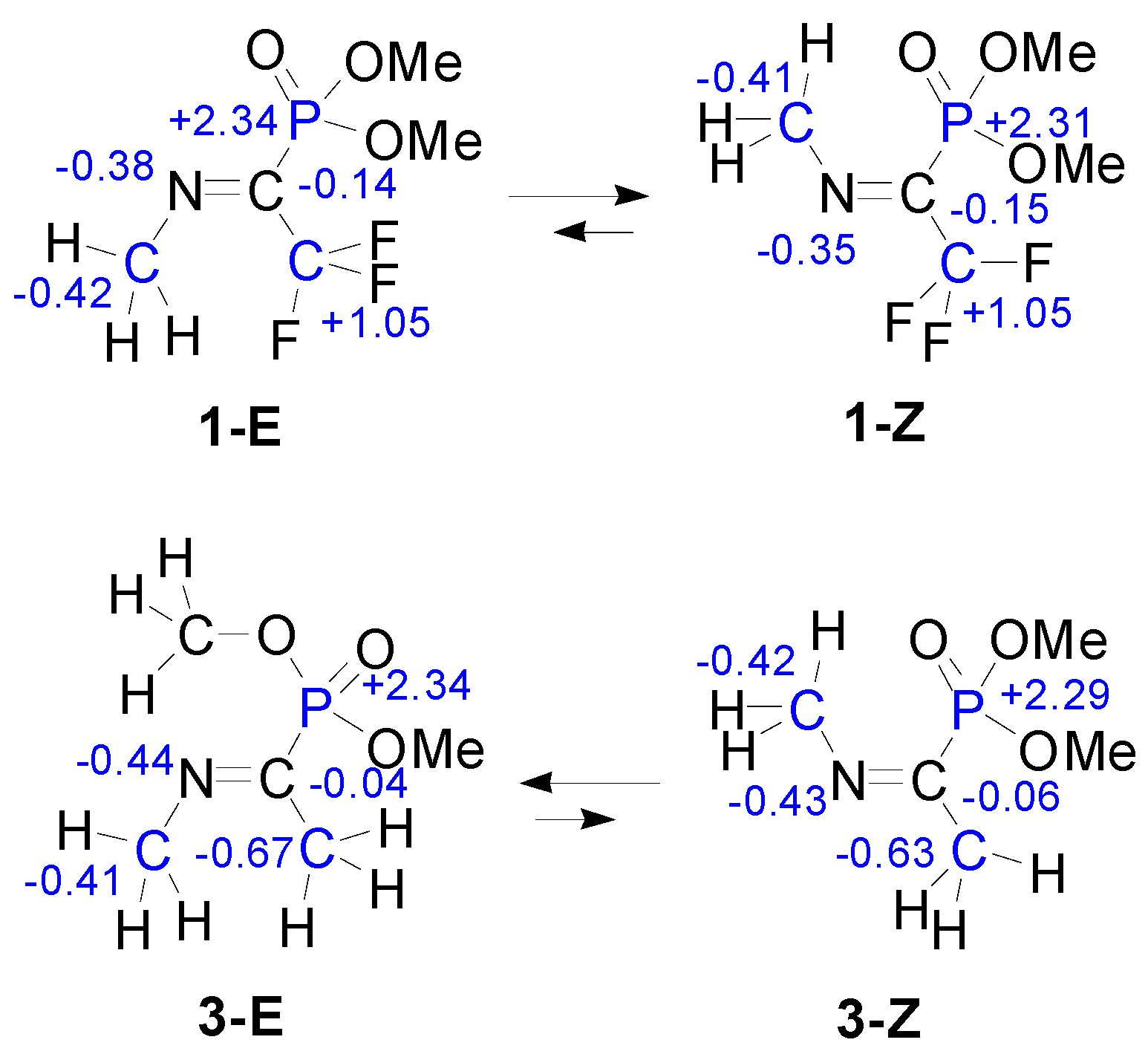{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
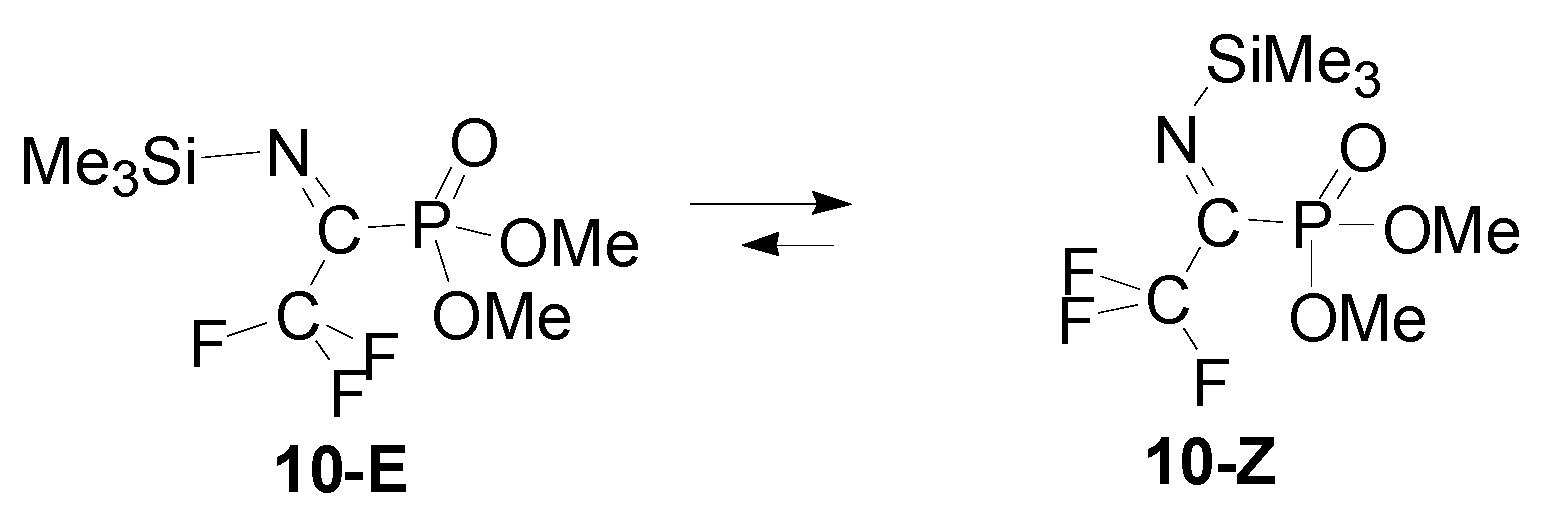{kind=link}
{kind=link}
| Entry | R1 | R2 | R3 | Z/E | ΔG(Z/E) a |
|---|---|---|---|---|---|
| 1 | H | CF3 | Et | 10:1 [12] | |
| 2 | H | CHF2 | Et | 5:1 | |
| 3 | H | C3F7 | Et | 20:1 | |
| 4 | Ph | CF3 | Et | 7:1 | −16.0 |
| 5 | 4-MeOC6H4 | CF3 | Me | 5:1 [9] | −14.4 |
| 6 | 4-CNC6H4 | CF3 | Me | 7:1 [9] | −19.7 |
| 7 | Me | CF3 | Et | 10:1 | |
| 8 | cyc-Pr | CF3 | Et | 11:1 [11] | −5.9 |
| 9 | Me | Ph | Et | 1:17 [9] | |
| 10 | CHMe2 | Ph | Et | 1:20 | |
| 11 | 4-MeOC6H4 | Ph | Et | 1:13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozhenko, A.B.; Kyrylchuk, A.A.; Lapinska, Y.O.; Rassukana, Y.V.; Trachevsky, V.V.; Pirozhenko, V.V.; Leszczynski, J.; Onysko, P.P. Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research. Organics 2021, 2, 84-97. https://doi.org/10.3390/org2020008
Rozhenko AB, Kyrylchuk AA, Lapinska YO, Rassukana YV, Trachevsky VV, Pirozhenko VV, Leszczynski J, Onysko PP. Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research. Organics. 2021; 2(2):84-97. https://doi.org/10.3390/org2020008
Chicago/Turabian StyleRozhenko, Alexander B., Andrey A. Kyrylchuk, Yuliia O. Lapinska, Yuliya V. Rassukana, Vladimir V. Trachevsky, Volodymyr V. Pirozhenko, Jerzy Leszczynski, and Petro P. Onysko. 2021. "Z,E-Isomerism in a Series of Substituted Iminophosphonates: Quantum Chemical Research" Organics 2, no. 2: 84-97. https://doi.org/10.3390/org2020008