
Esterifications with 2-(Trimethylsilyl)ethyl 2,2,2-Trichloroacetimidate



Abstract
:1. Introduction
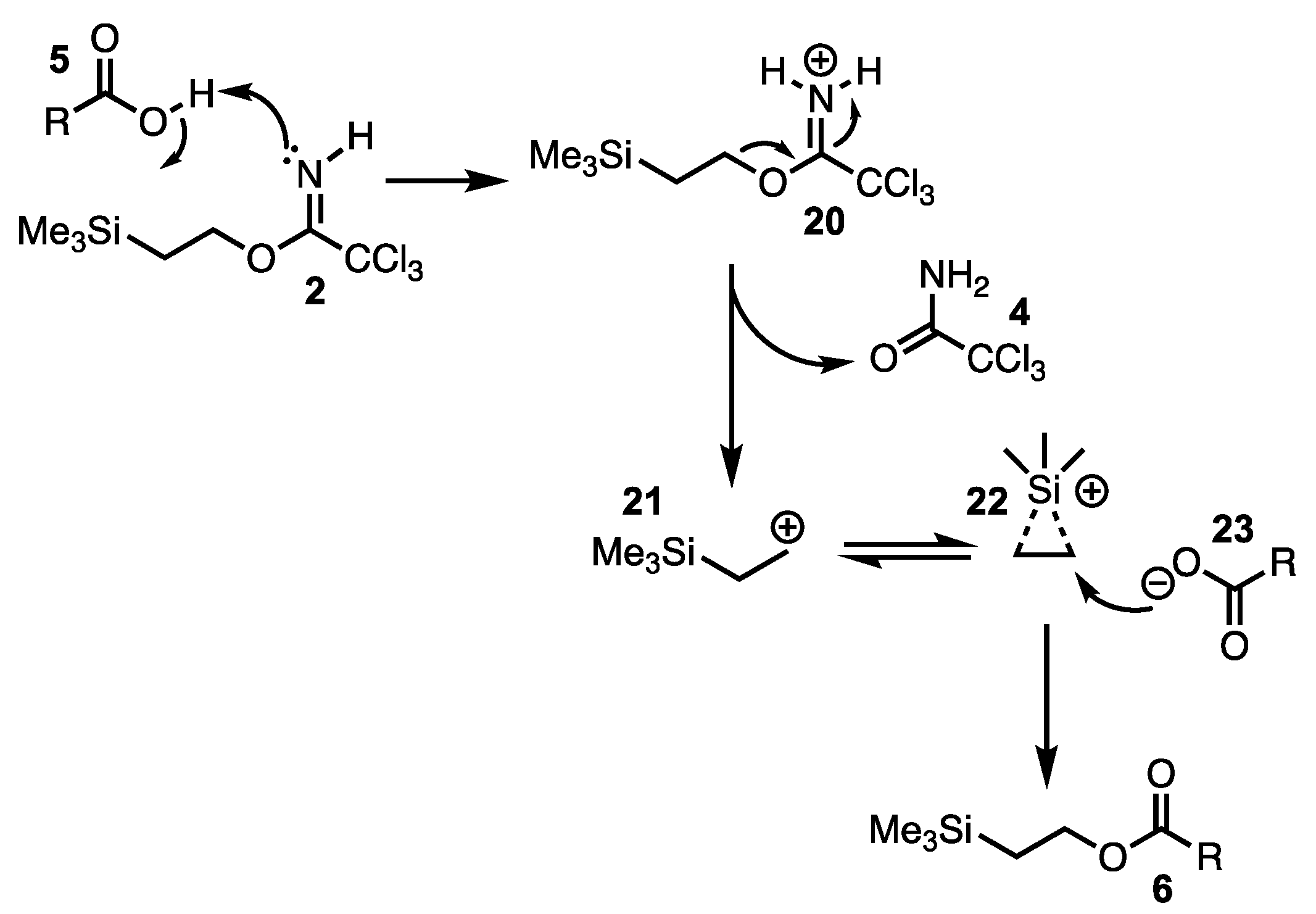
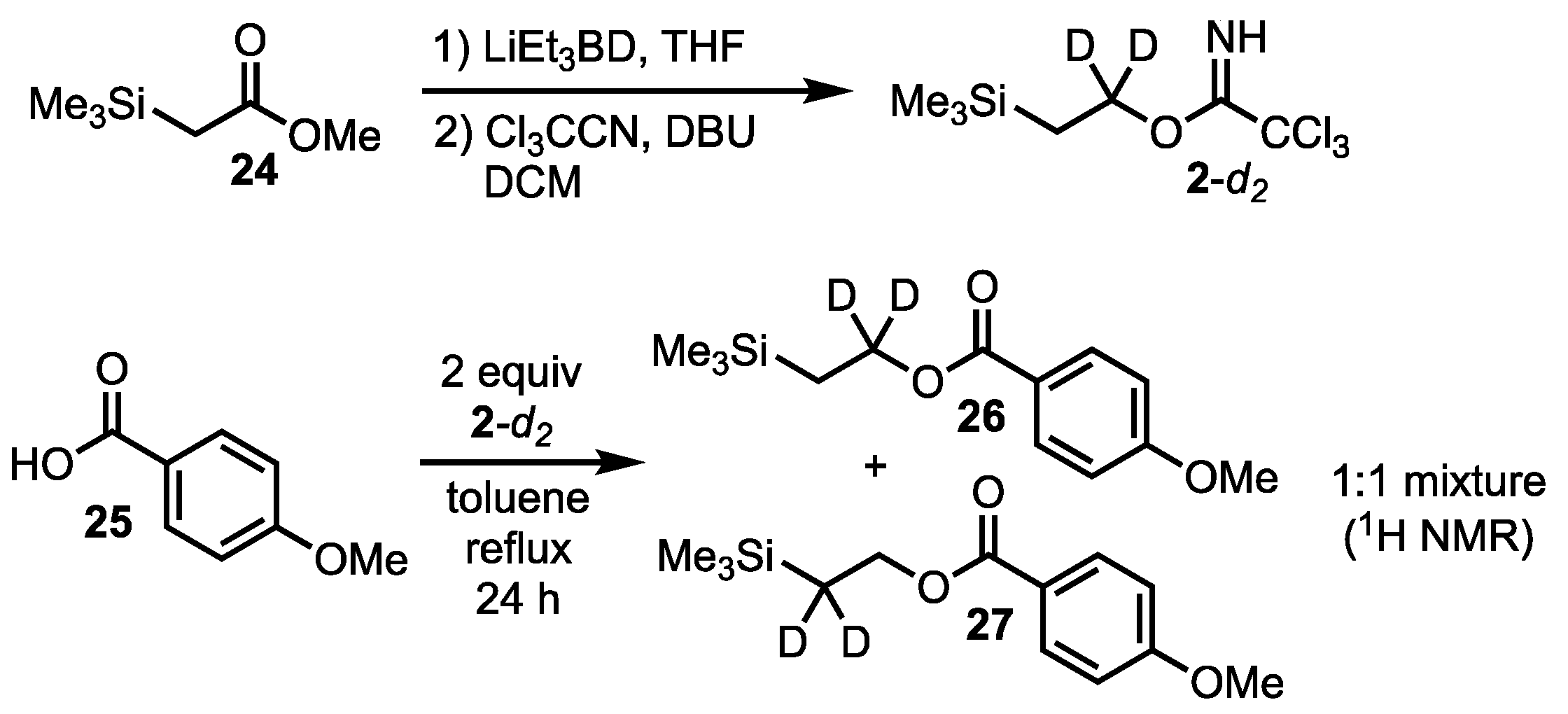
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Synthesis of Imidates
4.2. Esterification Reactions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wuts, P.G.M. Greene’s Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Kocienski, P.J. Protecting Groups, 3rd ed.; Thieme: Stuttgart, Germany, 2005. [Google Scholar]
- Sieber, P. The 2-trimethylsilylethyl residue, a selectively cleavable carboxyl protecting group. Helv. Chim. Acta 1977, 60, 2711–2716. [Google Scholar] [CrossRef]
- Gerlach, H. 2-(Trimethylsilyl)ethyl esters as a carboxyl protecting group; application in the synthesis of (-)-(S)-curvularin. Helv. Chim. Acta 1977, 60, 3039–3044. [Google Scholar] [CrossRef]
- Wood, J.L.; Thompson, B.D.; Yusuff, N.; Pflum, D.A.; Matthaeus, M.S.P. Total Synthesis of (±)-Epoxysorbicillinol. J. Am. Chem. Soc. 2001, 123, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Wu, M.H.; Regueiro-Ren, A.; St. Laurent, D.R.; Carroll, T.M.; Balasubramanian, B.N. Mild deprotection of 2-(trimethylsilyl)ethyl esters. Tetrahedron Lett. 2001, 42, 8593–8595. [Google Scholar] [CrossRef]
- Allen, J.G.; Danishefsky, S.J. The Total Synthesis of (±)-Rishirilide B. J. Am. Chem. Soc. 2001, 123, 351–352. [Google Scholar] [CrossRef]
- Forsch, R.A.; Rosowsky, A. Methotrexate analogs. 21. Selective cleavage of 2-(trimethylsilyl)ethyl esters in the presence of N-tert-butyloxycarbonyl groups during the synthesis of protected dilysine and trilysine derivatives of methotrexate. J. Org. Chem. 1984, 49, 1305–1309. [Google Scholar] [CrossRef]
- Fustero, S.; Sancho, A.G.; Acena, J.L.; Sanz-Cervera, J.F. Fluorous TBAF: A Convenient and Selective Reagent for Fluoride-Mediated Deprotections. J. Org. Chem. 2009, 74, 6398–6401. [Google Scholar] [CrossRef]
- Vedejs, E.; Larsen, S.D. Total synthesis of d,l-fulvine and d,l-crispatine. J. Am. Chem. Soc. 1984, 106, 3030–3032. [Google Scholar] [CrossRef]
- Fustero, S.; Sanchez-Rosello, M.; Rodrigo, V.; Garcia, A.; Catalan, S.; del Pozo, C. Selective Formal Transesterification of Fluorinated 2-(Trimethylsilyl)ethyl α-Imino Esters Mediated by TBAF. J. Org. Chem. 2008, 73, 5617–5620. [Google Scholar] [CrossRef]
- Reeves, C.M.; Behenna, D.C.; Stoltz, B.M. Development of (Trimethylsilyl)ethyl Ester Protected Enolates and Applications in Palladium-Catalyzed Enantioselective Allylic Alkylation: Intermolecular Cross-Coupling of Functionalized Electrophiles. Org. Lett. 2014, 16, 2314–2317. [Google Scholar] [CrossRef]
- Bourne, G.T.; Horwell, D.C.; Pritchard, M.C. Synthesis of an α-CH2CO2H functionalized tryptophan and its incorporation into an analog of cholecystokinin. Tetrahedron 1991, 47, 4763–4774. [Google Scholar] [CrossRef]
- Boger, D.L.; Yohannes, D. Total synthesis of OF4949-III and OF4949-IV: Unusual effects of remote substituents on the rate of macrocyclization reactions. Tetrahedron Lett. 1989, 30, 5061–5064. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Namoto, K.; Ritzen, A.; Ulven, T.; Shoji, M.; Li, J.; D’Amico, G.; Liotta, D.; French, C.T.; Wartmann, M.; et al. Chemical synthesis and biological evaluation of cis- and trans-12,13-cyclopropyl and 12,13-cyclobutyl epothilones and related pyridine side chain analogues. J. Am. Chem. Soc. 2001, 123, 9313–9323. [Google Scholar] [CrossRef]
- Baran, P.S.; Shenvi, R.A. Total Synthesis of (±)-Chartelline C. J. Am. Chem. Soc. 2006, 128, 14028–14029. [Google Scholar] [CrossRef] [PubMed]
- Horning, B.D.; MacMillan, D.W.C. Nine-Step Enantioselective Total Synthesis of (-)-Vincorine. J. Am. Chem. Soc. 2013, 135, 6442–6445. [Google Scholar] [CrossRef] [Green Version]
- Narasaka, K.; Sakakura, T.; Uchimaru, T.; Guedin-Vuong, D. Total synthesis of a macrocyclic pyrrolizidine alkaloid, (±)-integerrimine, utilizing an activable protecting group. J. Am. Chem. Soc. 1984, 106, 2954–2961. [Google Scholar] [CrossRef]
- Knutsen, L.J.S.; Judkins, B.D.; Newton, R.F.; Scopes, D.I.C.; Klinkert, G. Synthesis of imidazo-fused bridgehead-nitrogen 2’-deoxyribo-C-nucleosides: Coupling-elimination reactions of 2,5-anhydro-3,4,6-tri-O-benzoyl-D-allonic acid. J. Chem. Soc. Perkin Trans. 1985, 1, 621–630. [Google Scholar] [CrossRef]
- White, J.D.; Amedio, J.C., Jr.; Gut, S.; Ohira, S.; Jayasinghe, L.R. Stereoselective synthesis of the pyrrolizidine alkaloids (-)-integerrimine and (+)-usaramine. J. Org. Chem. 1992, 57, 2270–2284. [Google Scholar] [CrossRef]
- Jones, P.; Villeneuve, G.B.; Fei, C.; De Marte, J.; Haggarty, A.J.; Nwe, K.T.; Martin, D.A.; Lebuis, A.-M.; Finkelstein, J.M.; Gour-Salin, B.J.; et al. Synthesis and Structure-Activity Relationships of 2-Pyrazinylcarboxamidobenzoates and β-Ionylideneacetamidobenzoates with Retinoidal Activity. J. Med. Chem. 1998, 41, 3062–3077. [Google Scholar] [CrossRef]
- Roush, W.R.; Madar, D.J.; Coffey, D.S. Synthesis of highly functionalized naphthoate precursors to damavaricin D—Observation of kinetically stable benzocyclohexadienones in the bromination reactions of highly functionalized β-naphthol derivatives. Can. J. Chem. 2001, 79, 1711–1726. [Google Scholar]
- Fürstner, A.; Bindl, M.; Jean, L. Concise total synthesis of cruentaren A. Angew. Chem. Int. Ed. 2007, 46, 9275–9278. [Google Scholar] [CrossRef]
- Reed, S.A.; White, M.C. Catalytic Intermolecular Linear Allylic C-H Amination via Heterobimetallic Catalysis. J. Am. Chem. Soc. 2008, 130, 3316–3318. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tang, S.; Guo, Y.; Ye, T. Total synthesis of anti-tuberculosis natural products Ilamycins E1 and F. Org. Lett. 2018, 20, 6166–6169. [Google Scholar] [CrossRef]
- Wallach, D.R.; Stege, P.C.; Shah, J.P.; Chisholm, J.D. Brønsted Acid Catalyzed Monoalkylation of Anilines with Trichloroacetimidates. J. Org. Chem. 2015, 80, 1993–2000. [Google Scholar] [CrossRef]
- Wallach, D.R.; Chisholm, J.D. Alkylation of Sulfonamides with Trichloroacetimidates under Thermal Conditions. J. Org. Chem. 2016, 81, 8035–8042. [Google Scholar] [CrossRef] [PubMed]
- Duffy, B.C.; Howard, K.T.; Chisholm, J.D. Alkylation of thiols with trichloroacetimidates under neutral conditions. Tetrahedron Lett. 2015, 56, 3301–3305. [Google Scholar] [CrossRef]
- Howard, K.T.; Duffy, B.C.; Linaburg, M.R.; Chisholm, J.D. Formation of DPM ethers using O-diphenylmethyl trichloroacetimidate under thermal conditions. Org. Biomol. Chem. 2016, 14, 1623–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierry, J.; Yue, C.; Potier, P. 2-Phenylisopropyl and t-butyl trichloroacetimidates: Useful reagents for ester preparation of N-protected amino acids under neutral conditions. Tetrahedron Lett. 1998, 39, 1557–1560. [Google Scholar] [CrossRef]
- Respondek, T.; Cueny, E.; Kodanko, J.J. Cumyl Ester as the C-Terminal Protecting Group in the Enantioselective Alkylation of Glycine Benzophenone Imine. Org. Lett. 2012, 14, 150–153. [Google Scholar] [CrossRef]
- Shah, J.P.; Russo, C.M.; Howard, K.T.; Chisholm, J.D. Spontaneous formation of PMB esters using 4-methoxybenzyl-2,2,2-trichloroacetimidate. Tetrahedron Lett. 2014, 55, 1740–1742. [Google Scholar] [CrossRef]
- Adhikari, A.A.; Shah, J.P.; Howard, K.T.; Russo, C.M.; Wallach, D.R.; Linaburg, M.R.; Chisholm, J.D. Convenient Formation of Diphenylmethyl Esters Using Diphenylmethyl Trichloroacetimidate. Synlett 2014, 25, 283–287. [Google Scholar]
- Mahajani, N.S.; Meador, R.I.L.; Smith, T.J.; Canarelli, S.E.; Adhikari, A.A.; Shah, J.P.; Russo, C.M.; Wallach, D.R.; Howard, K.T.; Millimaci, A.M.; et al. Ester Formation via Symbiotic Activation Utilizing Trichloroacetimidate Electrophiles. J. Org. Chem. 2019, 84, 7871–7882. [Google Scholar] [CrossRef]
- Ali, I.A.I.; Ashry, E.S.H.E.; Schmidt, R.R. Protection of Hydroxy Groups with Diphenylmethyl and 9-Fluorenyl Trichloroacetimidates—Effect on Anomeric Stereocontrol. Eur. J. Org. Chem. 2003, 2003, 4121–4131. [Google Scholar] [CrossRef]
- Green, A.J.; Kuan, Y.L.; White, J.M. Rearrangement of r-5-methyl-c-2-(trimethylsilyl)cyclohexan-t-yl 2,4-dinitrobenzoate in chloroform. J. Chem. Soc. Chem. Commun. 1994, 2023–2024. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Effenberger, G. O-glycosyl imidates. 29. Reaction of O-(glucopyranosyl) imidates with electron-rich heterocycles. Synthesis of C-glucosides. Liebigs Ann. Chem. 1987, 19, 825–831. [Google Scholar] [CrossRef]
- Li, C.; Wang, J. Lewis Acid Catalyzed Propargylation of Arenes with O-Propargyl Trichloroacetimidates: Synthesis of 1,3-Diarylpropynes. J. Org. Chem. 2007, 72, 7431–7434. [Google Scholar] [CrossRef] [PubMed]
- Devineau, A.; Pousse, G.; Taillier, C.; Blanchet, J.; Rouden, J.; Dalla, V. One-Pot Hydroxy Group Activation/Carbon-Carbon Bond Forming Sequence Using a Brønsted Base/Brønsted Acid System. Adv. Synth. Catal. 2010, 352, 2881–2886. [Google Scholar] [CrossRef]
- Wiebe, C.; de la Sotilla, S.F.; Opatz, T. Synthesis of 1,3- and 2,3-diglycosylated indoles as potential trisaccharide mimetics. Synthesis 2012, 44, 1385–1397. [Google Scholar]
- Adhikari, A.A.; Radal, L.; Chisholm, J.D. Synthesis of 3,3’-Disubstituted Indolenines Utilizing the Lewis Acid Catalyzed Alkylation of 2,3-Disubstituted Indoles with Trichloroacetimidates. Synlett 2017, 28, 2335–2339. [Google Scholar] [PubMed]
- Suzuki, T.; Chisholm, J.D. Friedel-Crafts alkylation of indoles with trichloroacetimidates. Tetrahedron Lett. 2019, 60, 1325–1329. [Google Scholar] [CrossRef]
- Lambert, J.B.; Wang, G.T.; Finzel, R.B.; Teramura, D.H. Stabilization of positive charge by β-silicon. J. Am. Chem. Soc. 1987, 109, 7838–7845. [Google Scholar] [CrossRef]
- Chiavarino, B.; Crestoni, M.E.; Lemaire, J.; Maitre, P.; Fornarini, S. Communication: Infrared spectroscopy of protonated allyltrimethylsilane: Evidence for the β-silyl effect. J. Chem. Phys. 2013, 139, 071102. [Google Scholar] [CrossRef]
- Sharma, P.K.; Dawid, M.; Warkentin, J.; Vestal, R.M.; Wudl, F. Mechanism of Migration of the Trimethylsilyl Group during Reactions of Methoxy[(trimethylsilyl)ethoxy]carbene with N-Phenylmaleimide and C60. J. Org. Chem. 2001, 66, 7496–7499. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Issa, W.; White, J.M. Influence of trimethylsilyl substituents on primary C-O bond lengths at the β-position: Results from low-temperature x-ray structural studies. Aust. J. Chem. 1997, 50, 927–932. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}

| Entry | Equiv 2 | Promoter (Equiv.) | Solvent | t (h) | T | Yield a |
|---|---|---|---|---|---|---|
| 1 | 1.2 | TMSOTf (1.0) | DCM | 1 | rt | 0 b |
| 2 | 1.2 | TMSOTf (0.2) | DCM | 1 | rt | 0 b |
| 3 | 1.2 | CSA (1.0) | DCM | 1 | rt | 0 b |
| 4 | 1.2 | CSA (0.2) | DCM | 1 | rt | 22 |
| 5 | 1.2 | PPTS (1.0) | DCM | 1 | rt | 0 b |
| 6 | 1.2 | PPTS (0.2) | DCM | 1 | rt | 17 |
| 7 | 2.0 | none | toluene | 24 | reflux | 81 c |

| Entry | Product | Yield |
|---|---|---|
| 1 |  | 81 |
| 2 |  | 56 |
| 3 |  | 66 |
| 4 |  | 58 |
| 5 |  | 56 |
| 6 |  | 90 |
| 7 |  | 65 |
| 8 |  | 44 a |
| 9 |  | 75 |
| 10 |  | 76 |
| 11 |  | 63 |
| 12 |  | 57 |
| 13 |  | 45 b |
| 14 |  | 38 (78 c) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Meyer, S.T.; Dormann, S.; Chisholm, J.D. Esterifications with 2-(Trimethylsilyl)ethyl 2,2,2-Trichloroacetimidate. Organics 2021, 2, 17-25. https://doi.org/10.3390/org2010002
Lin W, Meyer ST, Dormann S, Chisholm JD. Esterifications with 2-(Trimethylsilyl)ethyl 2,2,2-Trichloroacetimidate. Organics. 2021; 2(1):17-25. https://doi.org/10.3390/org2010002
Chicago/Turabian StyleLin, Wenhong, Shea T. Meyer, Shawn Dormann, and John D. Chisholm. 2021. "Esterifications with 2-(Trimethylsilyl)ethyl 2,2,2-Trichloroacetimidate" Organics 2, no. 1: 17-25. https://doi.org/10.3390/org2010002